

Molecular mechanisms of HPV mediated neoplastic progression
- PMID: 27933097
- PMCID: PMC5123406
- DOI: 10.1186/s13027-016-0107-4
Molecular mechanisms of HPV mediated neoplastic progression
Abstract
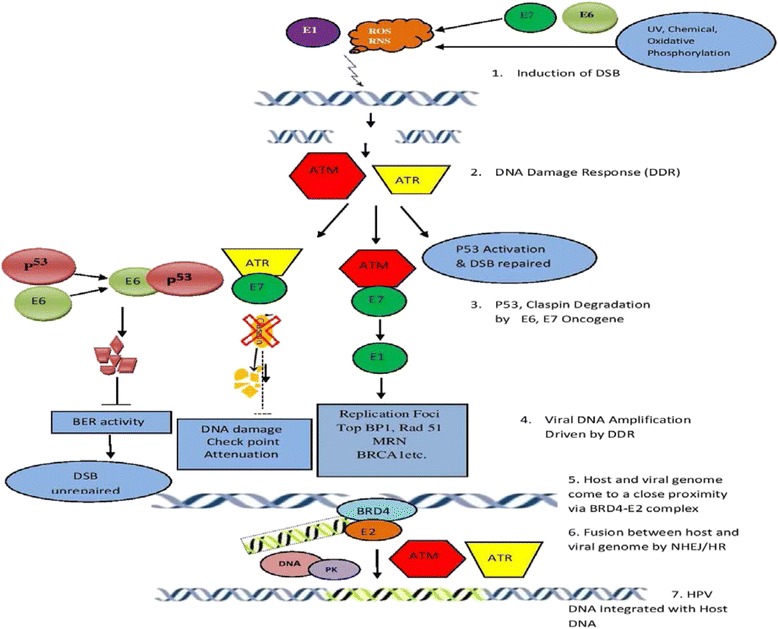
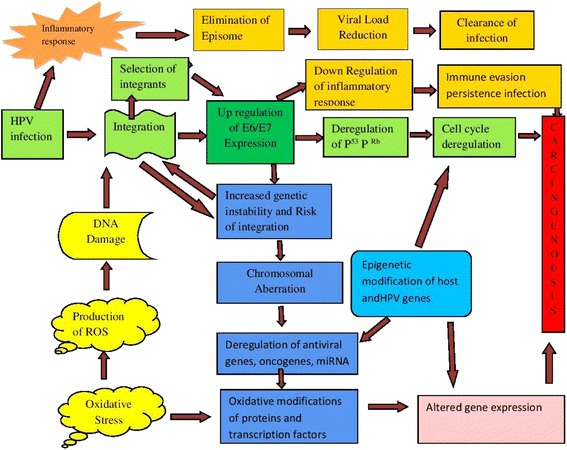
Human Papillomavirus is the major etiological agent in the development of cervical cancer but not a sufficient cause. Despite significant research, the underlying mechanisms of progression from a low-grade squamous intraepithelial lesion to high grade squamous intraepithelial lesion are yet to be understood. Deregulation of viral gene expression and host genomic instability play a central role in virus-mediated carcinogenesis. Key events such as viral integration and epigenetic modifications may lead to the deregulation of viral and host gene expression. This review has summarized the available literature to describe the possible mechanism and role of viral integration in mediating carcinogenesis. HPV integration begins with DNA damage or double strand break induced either by oxidative stress or HPV proteins and the subsequent steps are driven by the DNA damage responses. Inflammation and oxidative stress could be considered as cofactors in stimulating viral integration and deregulation of cellular and viral oncogenes during the progression of cervical carcinoma. All these events together with the host and viral genetic and epigenetic modifications in neoplastic progression have also been reviewed which may be relevant in identifying a new preventive therapeutic strategy. In the absence of therapeutic intervention for HPV-infected individuals, future research focus should be directed towards preventing and reversing of HPV integration. DNA damage response, knocking out integrated HPV sequences, siRNA approach, modulating the selection mechanism of cells harboring integrated genomes and epigenetic modifiers are the possible therapeutic targets.
Keywords: Carcinogenesis; Cervical cancer; HPV integration; Neoplastic progression.
Figures
References
-
- Cervical cancer: estimated incidence mortality and prevalence worldwide in 2012. Globacan 2012. “http://globocan.iarc.fr/old/FactSheets/cancers/cervix-new.asp. Accessed 5 Dec 2015.
-
- Howley PM, Lowry DR. Papillomaviruses. In: Knipe DM and Howley PM editors Fields virology. Philadelphia: Lippincott, Williams and Wilkins; 2007. p. 2299-354
-
- Guan P, Howell-Jones R, Li N, Bruni L, et al. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer Int. J Cancer. 2012;131:2349–2359. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
