

High-throughput allele-specific expression across 250 environmental conditions
- PMID: 27934696
- PMCID: PMC5131815
- DOI: 10.1101/gr.209759.116
High-throughput allele-specific expression across 250 environmental conditions
Abstract
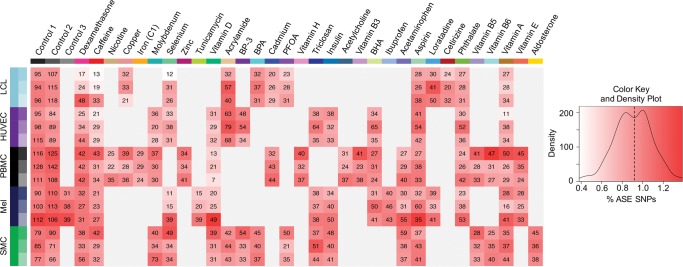
Gene-by-environment (GxE) interactions determine common disease risk factors and biomedically relevant complex traits. However, quantifying how the environment modulates genetic effects on human quantitative phenotypes presents unique challenges. Environmental covariates are complex and difficult to measure and control at the organismal level, as found in GWAS and epidemiological studies. An alternative approach focuses on the cellular environment using in vitro treatments as a proxy for the organismal environment. These cellular environments simplify the organism-level environmental exposures to provide a tractable influence on subcellular phenotypes, such as gene expression. Expression quantitative trait loci (eQTL) mapping studies identified GxE interactions in response to drug treatment and pathogen exposure. However, eQTL mapping approaches are infeasible for large-scale analysis of multiple cellular environments. Recently, allele-specific expression (ASE) analysis emerged as a powerful tool to identify GxE interactions in gene expression patterns by exploiting naturally occurring environmental exposures. Here we characterized genetic effects on the transcriptional response to 50 treatments in five cell types. We discovered 1455 genes with ASE (FDR < 10%) and 215 genes with GxE interactions. We demonstrated a major role for GxE interactions in complex traits. Genes with a transcriptional response to environmental perturbations showed sevenfold higher odds of being found in GWAS. Additionally, 105 genes that indicated GxE interactions (49%) were identified by GWAS as associated with complex traits. Examples include GIPR-caffeine interaction and obesity and include LAMP3-selenium interaction and Parkinson disease. Our results demonstrate that comprehensive catalogs of GxE interactions are indispensable to thoroughly annotate genes and bridge epidemiological and genome-wide association studies.
© 2016 Moyerbrailean et al.; Published by Cold Spring Harbor Laboratory Press.
Figures





References
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 57: 289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases