

Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: a novel strategy to treat acute gout
- PMID: 27934918
- PMCID: PMC5146947
- DOI: 10.1038/srep38622
Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: a novel strategy to treat acute gout
Abstract
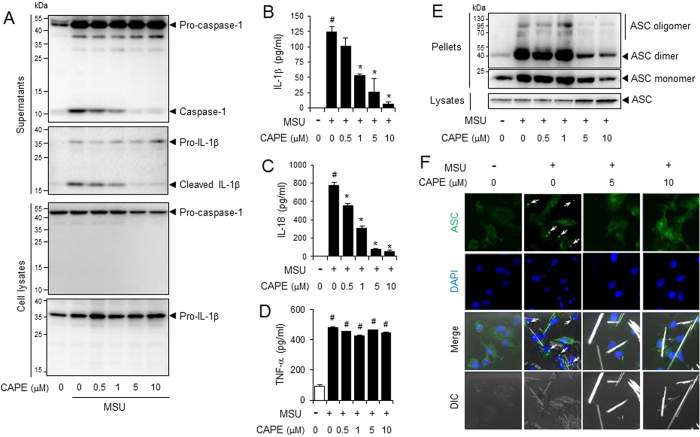
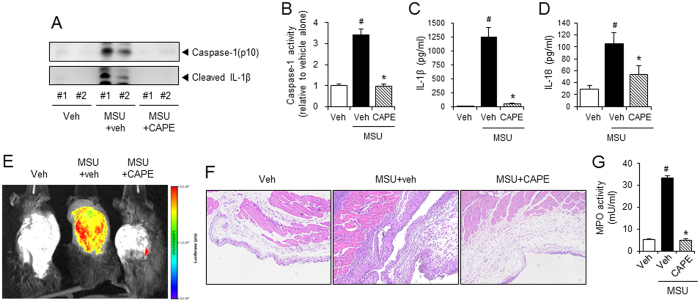
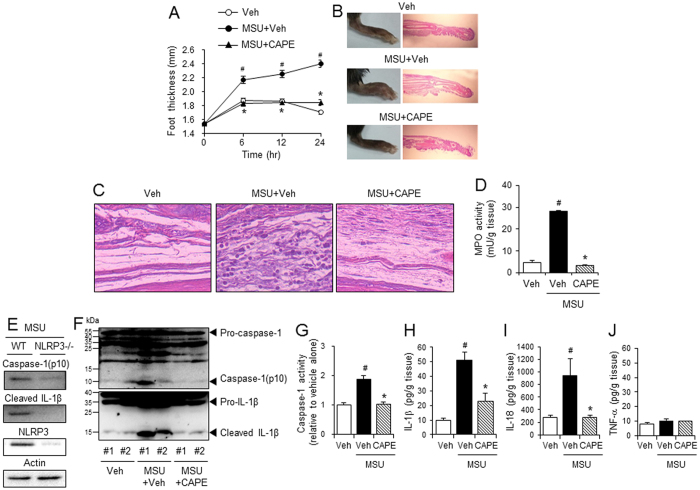
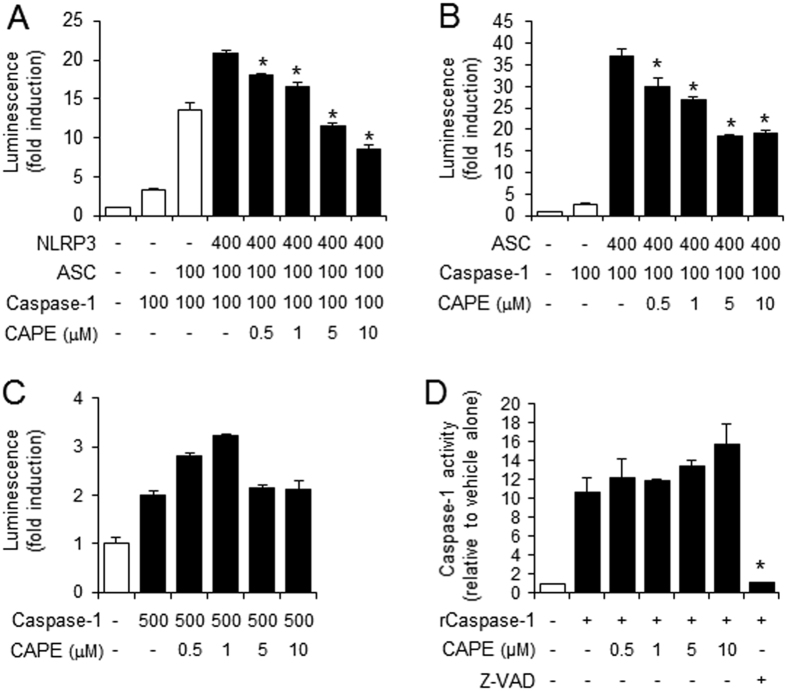
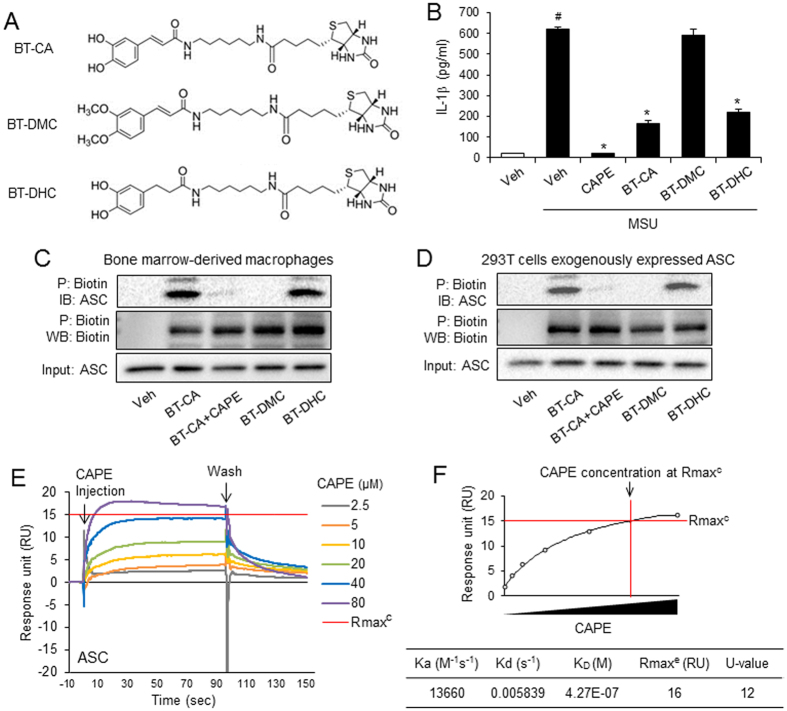
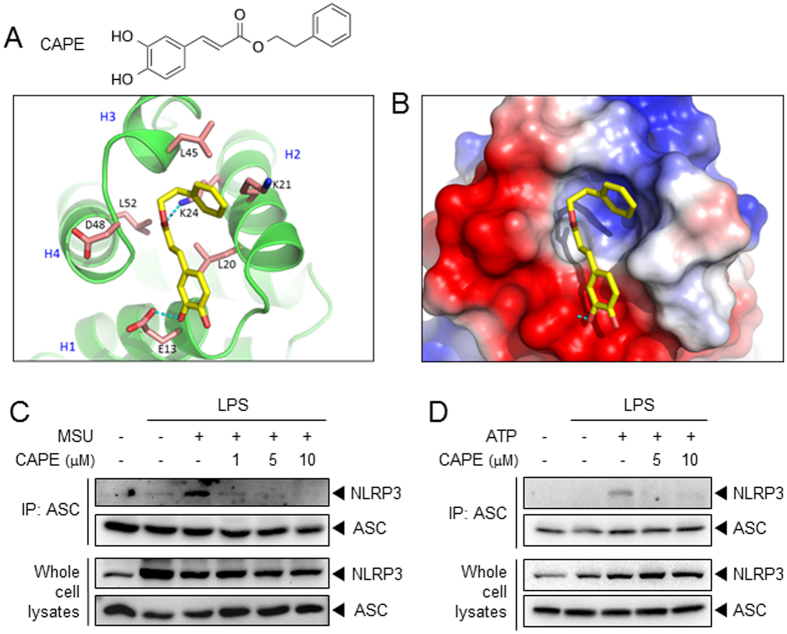
Gouty arthritis is caused by the deposition of uric acid crystals, which induce the activation of NOD-like receptor family, pyrin domain containing 3(NLRP3) inflammasome. The NLRP3 inflammasome, composed of NLRP3, the adaptor protein ASC, and caspase-1, is closely linked to the pathogenesis of various metabolic diseases including gouty arthritis. We investigated whether an orally administrable inhibitor of NLRP3 inflammasome was effective for alleviating the pathological symptoms of gouty arthritis and what was the underlying mechanism. In primary mouse macrophages, caffeic acid phenethyl ester(CAPE) blocked caspase-1 activation and IL-1β production induced by MSU crystals, showing that CAPE suppresses NLRP3 inflammasome activation. In mouse gouty arthritis models, oral administration of CAPE suppressed MSU crystals-induced caspase-1 activation and IL-1β production in the air pouch exudates and the foot tissues, correlating with attenuation of inflammatory symptoms. CAPE directly associated with ASC as shown by SPR analysis and co-precipitation, resulting in blockade of NLRP3-ASC interaction induced by MSU crystals. Our findings provide a novel regulatory mechanism by which small molecules harness the activation of NLRP3 inflammasome by presenting ASC as a new target. Furthermore, the results suggest the preventive or therapeutic strategy for NLRP3-related inflammatory diseases such as gouty arthritis using orally available small molecules.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
