

Inhibition of nonsense-mediated RNA decay by ER stress
- PMID: 27940503
- PMCID: PMC5311500
- DOI: 10.1261/rna.058040.116
Inhibition of nonsense-mediated RNA decay by ER stress
Abstract
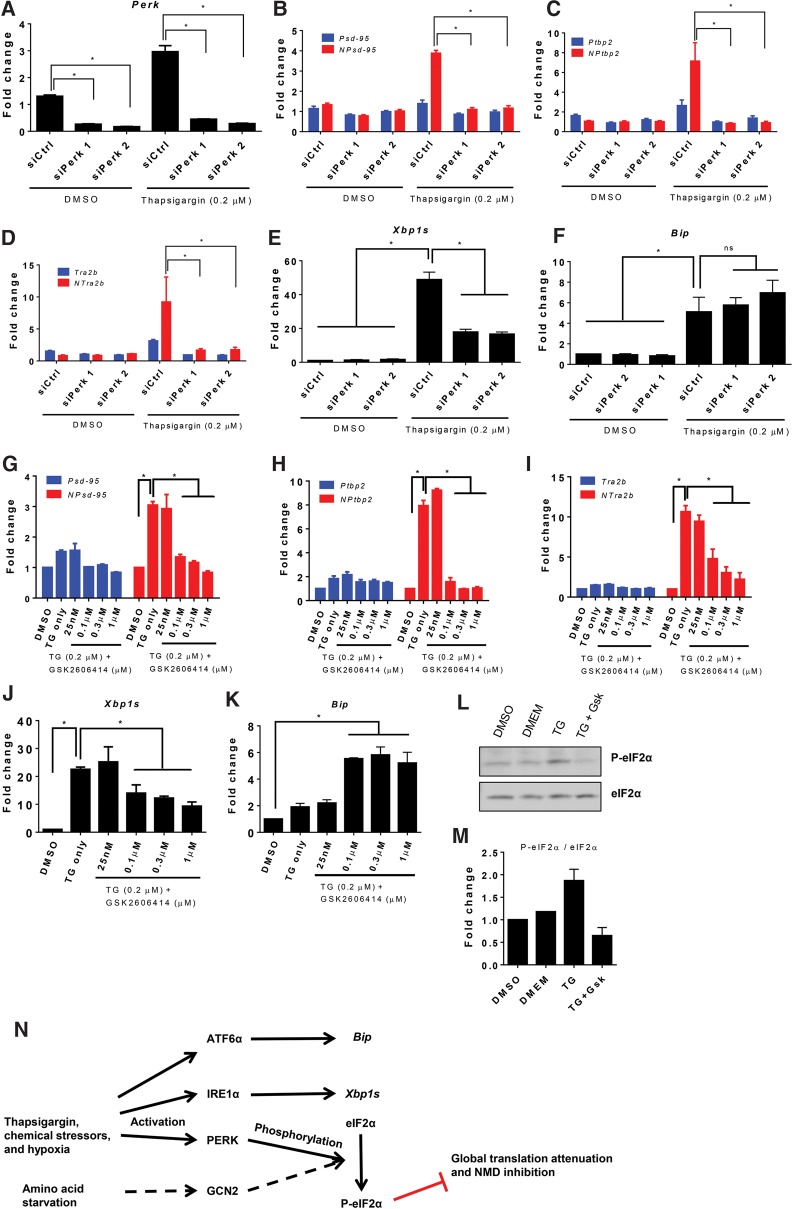
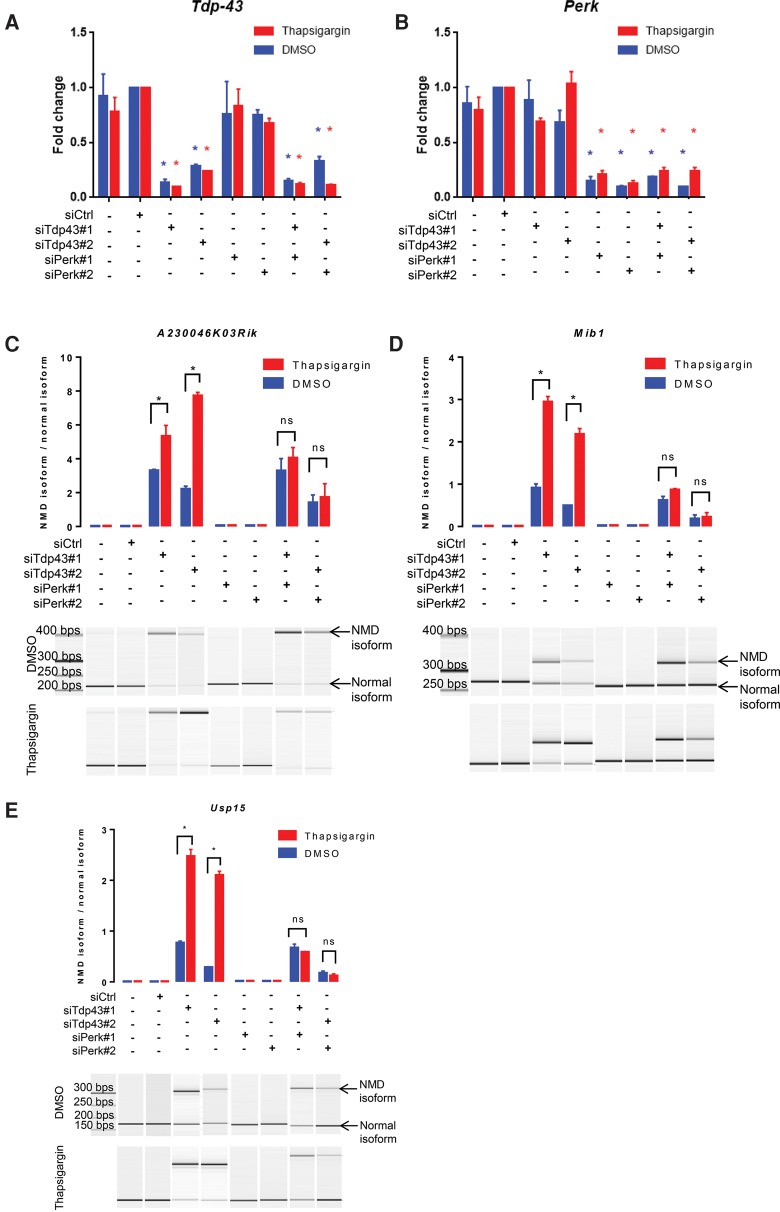
Nonsense-mediated RNA decay (NMD) selectively degrades mutated and aberrantly processed transcripts that contain premature termination codons (PTC). Cellular NMD activity is typically assessed using exogenous PTC-containing reporters. We overcame some inherently problematic aspects of assaying endogenous targets and developed a broadly applicable strategy to reliably and easily monitor changes in cellular NMD activity. Our new method was genetically validated for distinguishing NMD regulation from transcriptional control and alternative splicing regulation, and unexpectedly disclosed a different sensitivity of NMD targets to NMD inhibition. Applying this robust method for screening, we identified NMD-inhibiting stressors but also found that NMD inactivation was not universal to cellular stresses. The high sensitivity and broad dynamic range of our method revealed a strong correlation between NMD inhibition, endoplasmic reticulum (ER) stress, and polysome disassembly upon thapsigargin treatment in a temporal and dose-dependent manner. We found little evidence of calcium signaling mediating thapsigargin-induced NMD inhibition. Instead, we discovered that of the three unfolded protein response (UPR) pathways activated by thapsigargin, mainly protein kinase RNA-like endoplasmic reticulum kinase (PERK) was required for NMD inhibition. Finally, we showed that ER stress compounded TDP-43 depletion in the up-regulation of NMD isoforms that had been implicated in the pathogenic mechanisms of amyotrophic lateral sclerosis and frontotemporal dementia, and that the additive effect of ER stress was completely blocked by PERK deficiency.
Keywords: ALS; ATF6α; FTD; Hnrnpl; IREα; NMD; PERK; Psd-95; Ptbp1; Ptbp2; Srsf11; Tdp-43; Tra2b; UPR; Upf2; alternative splicing; cellular stress; cryptic splicing.
© 2017 Li et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
Similar articles
-
PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2.Nat Neurosci. 2012 Jan 15;15(3):381-8, S1. doi: 10.1038/nn.3026. Nat Neurosci. 2012. PMID: 22246437 Free PMC article.
-
Moderate endoplasmic reticulum stress activates a PERK and p38-dependent apoptosis.Cell Stress Chaperones. 2017 Jan;22(1):43-54. doi: 10.1007/s12192-016-0740-2. Epub 2016 Oct 20. Cell Stress Chaperones. 2017. PMID: 27761878 Free PMC article.
-
Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis.Cell Death Dis. 2020 May 26;11(5):397. doi: 10.1038/s41419-020-2601-2. Cell Death Dis. 2020. PMID: 32457286 Free PMC article.
-
Targeting ER stress/ER stress response in myopathies.Redox Biol. 2019 Sep;26:101232. doi: 10.1016/j.redox.2019.101232. Epub 2019 Jun 4. Redox Biol. 2019. PMID: 31181458 Free PMC article. Review.
-
Targeting UPR branches, a potential strategy for enhancing efficacy of cancer chemotherapy.Acta Biochim Biophys Sin (Shanghai). 2021 Nov 10;53(11):1417-1427. doi: 10.1093/abbs/gmab131. Acta Biochim Biophys Sin (Shanghai). 2021. PMID: 34664059 Review.
Cited by
-
Targeting nonsense-mediated RNA decay does not increase progranulin levels in the Grn R493X mouse model of frontotemporal dementia.PLoS One. 2023 Mar 9;18(3):e0282822. doi: 10.1371/journal.pone.0282822. eCollection 2023. PLoS One. 2023. PMID: 36893203 Free PMC article.
-
Developmental Xist induction is mediated by enhanced splicing.Nucleic Acids Res. 2019 Feb 20;47(3):1532-1543. doi: 10.1093/nar/gky1198. Nucleic Acids Res. 2019. PMID: 30496473 Free PMC article.
-
Selective and competitive functions of the AAR and UPR pathways in stress-induced angiogenesis.Cell Discov. 2021 Oct 26;7(1):98. doi: 10.1038/s41421-021-00332-8. Cell Discov. 2021. PMID: 34697290 Free PMC article.
-
Compromised nonsense-mediated RNA decay results in truncated RNA-binding protein production upon DUX4 expression.Cell Rep. 2023 Jun 27;42(6):112642. doi: 10.1016/j.celrep.2023.112642. Epub 2023 Jun 13. Cell Rep. 2023. PMID: 37314931 Free PMC article.
-
A WFS1 variant disrupting acceptor splice site uncovers the impact of alternative splicing on beta cell apoptosis in a patient with Wolfram syndrome.Diabetologia. 2025 Jan;68(1):128-151. doi: 10.1007/s00125-024-06307-0. Epub 2024 Nov 9. Diabetologia. 2025. PMID: 39520565 Free PMC article.
References
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. 2006. TDP-43 is a component of ubiquitin-positive τ-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611. - PubMed
-
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, et al. 2012. Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55: 7193–7207. - PubMed
-
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous