

Peroxisome biogenesis and human peroxisome-deficiency disorders
- PMID: 27941306
- PMCID: PMC5328784
- DOI: 10.2183/pjab.92.463
Peroxisome biogenesis and human peroxisome-deficiency disorders
Abstract
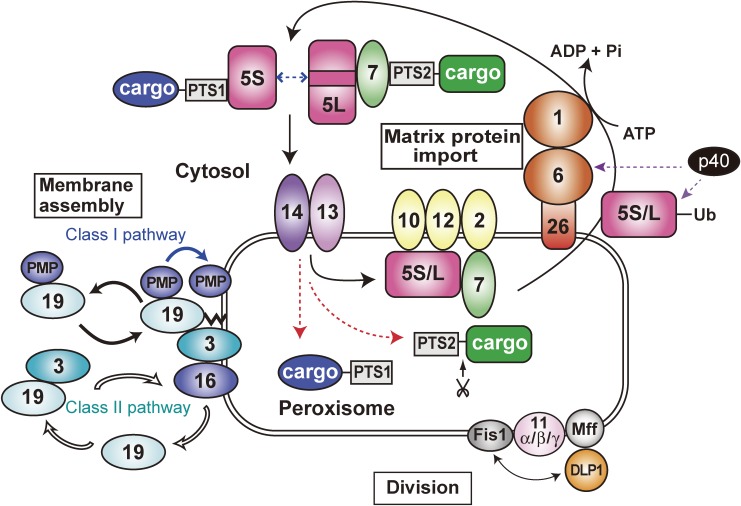
Peroxisome is a single-membrane-bounded ubiquitous organelle containing a hundred different enzymes that catalyze various metabolic pathways such as β-oxidation of very long-chain fatty acids and synthesis of plasmalogens. To investigate peroxisome biogenesis and human peroxisome biogenesis disorders (PBDs) including Zellweger syndrome, more than a dozen different complementation groups of Chinese hamster ovary (CHO) cell mutants impaired in peroxisome biogenesis are isolated as a model experimental system. By taking advantage of rapid functional complementation assay of the CHO cell mutants, successful cloning of PEX genes encoding peroxins required for peroxisome assembly invaluably contributed to the accomplishment of cloning of pathogenic genes responsible for PBDs. Peroxins are divided into three groups: 1) peroxins including Pex3p, Pex16p and Pex19p, are responsible for peroxisome membrane biogenesis via Pex19p- and Pex3p-dependent class I and Pex19p- and Pex16p-dependent class II pathways; 2) peroxins that function in matrix protein import; 3) those such as Pex11pβ are involved in peroxisome division where DLP1, Mff, and Fis1 coordinately function.
Figures
Similar articles
-
Peroxisome biogenesis disorders: molecular basis for impaired peroxisomal membrane assembly: in metabolic functions and biogenesis of peroxisomes in health and disease.Biochim Biophys Acta. 2012 Sep;1822(9):1337-42. doi: 10.1016/j.bbadis.2012.06.004. Epub 2012 Jun 13. Biochim Biophys Acta. 2012. PMID: 22705440 Review.
-
Peroxisome biogenesis in mammalian cells.Front Physiol. 2014 Aug 15;5:307. doi: 10.3389/fphys.2014.00307. eCollection 2014. Front Physiol. 2014. PMID: 25177298 Free PMC article. Review.
-
Functional domain mapping of peroxin Pex19p: interaction with Pex3p is essential for function and translocation.J Cell Sci. 2006 Sep 1;119(Pt 17):3539-50. doi: 10.1242/jcs.03100. Epub 2006 Aug 8. J Cell Sci. 2006. PMID: 16895967
-
Generation of Peroxisome-Deficient Somatic Animal Cell Mutants.Methods Mol Biol. 2017;1595:319-327. doi: 10.1007/978-1-4939-6937-1_29. Methods Mol Biol. 2017. PMID: 28409474
-
Lessons from peroxisome-deficient Chinese hamster ovary (CHO) cell mutants.Biochim Biophys Acta. 2006 Dec;1763(12):1374-81. doi: 10.1016/j.bbamcr.2006.09.012. Epub 2006 Sep 14. Biochim Biophys Acta. 2006. PMID: 17045664 Review.
Cited by
-
The Cys-N-degron pathway modulates pexophagy through the N-terminal oxidation and arginylation of ACAD10.Autophagy. 2023 Jun;19(6):1642-1661. doi: 10.1080/15548627.2022.2126617. Epub 2022 Oct 2. Autophagy. 2023. PMID: 36184612 Free PMC article.
-
A Novel Mutation in PEX11β Gene.Iran J Child Neurol. 2021 Winter;15(1):93-100. doi: 10.22037/ijcn.v15i1.26129. Iran J Child Neurol. 2021. PMID: 33558817 Free PMC article.
-
Functional Analyses of a Putative, Membrane-Bound, Peroxisomal Protein Import Mechanism from the Apicomplexan Protozoan Toxoplasma gondii.Genes (Basel). 2018 Aug 29;9(9):434. doi: 10.3390/genes9090434. Genes (Basel). 2018. PMID: 30158461 Free PMC article.
-
Genotype-phenotype correlations and disease mechanisms in PEX13-related Zellweger spectrum disorders.Orphanet J Rare Dis. 2022 Jul 19;17(1):286. doi: 10.1186/s13023-022-02415-5. Orphanet J Rare Dis. 2022. PMID: 35854306 Free PMC article.
-
Lipid alterations in human frontal cortex in ALS-FTLD-TDP43 proteinopathy spectrum are partly related to peroxisome impairment.Neuropathol Appl Neurobiol. 2021 Jun;47(4):544-563. doi: 10.1111/nan.12681. Epub 2021 Jan 12. Neuropathol Appl Neurobiol. 2021. PMID: 33332650 Free PMC article.
References
-
- Wanders R.J.A., Waterham H.R. (2006) Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 75, 295–332. - PubMed
-
- de Duve C., Baudhuin P. (1966) Peroxisomes (microbodies and related particles). Physiol. Rev. 46, 323–357. - PubMed
-
- Lazarow P.B., Fujiki Y. (1985) Biogenesis of peroxisomes. Annu. Rev. Cell Biol. 1, 489–530. - PubMed
-
- Fujiki Y. (1997) Molecular defects in genetic diseases of peroxisomes. Biochim. Biophys. Acta 1361, 235–250. - PubMed
-
- Fujiki Y. (2000) Peroxisome biogenesis and peroxisome biogenesis disorders. FEBS Lett. 476, 42–46. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous