

Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia
- PMID: 27941792
- PMCID: PMC6548550
- DOI: 10.1038/nm.4247
Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia
Abstract
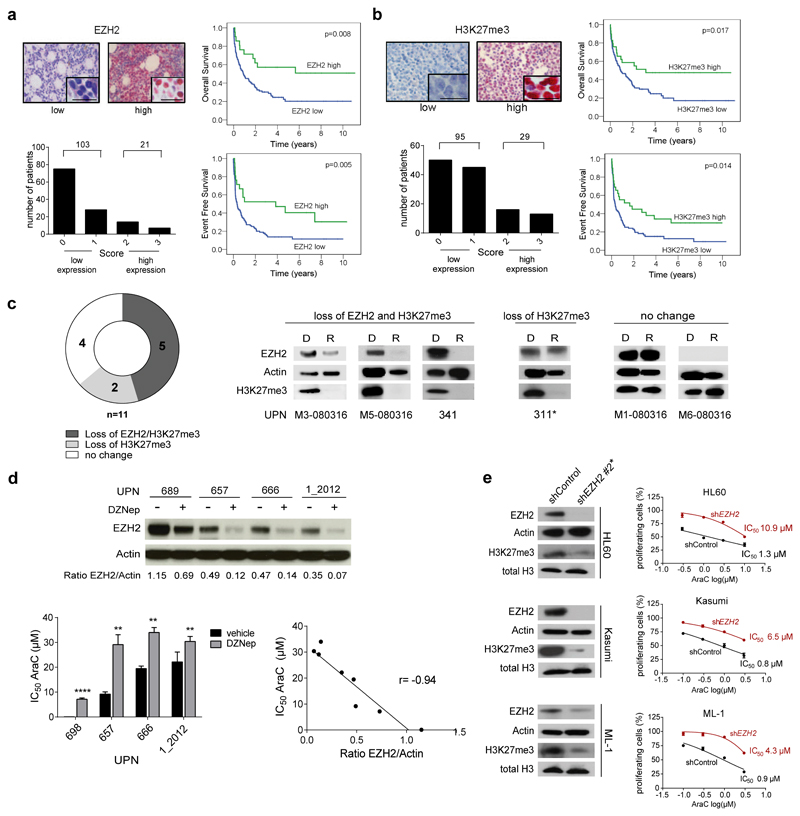
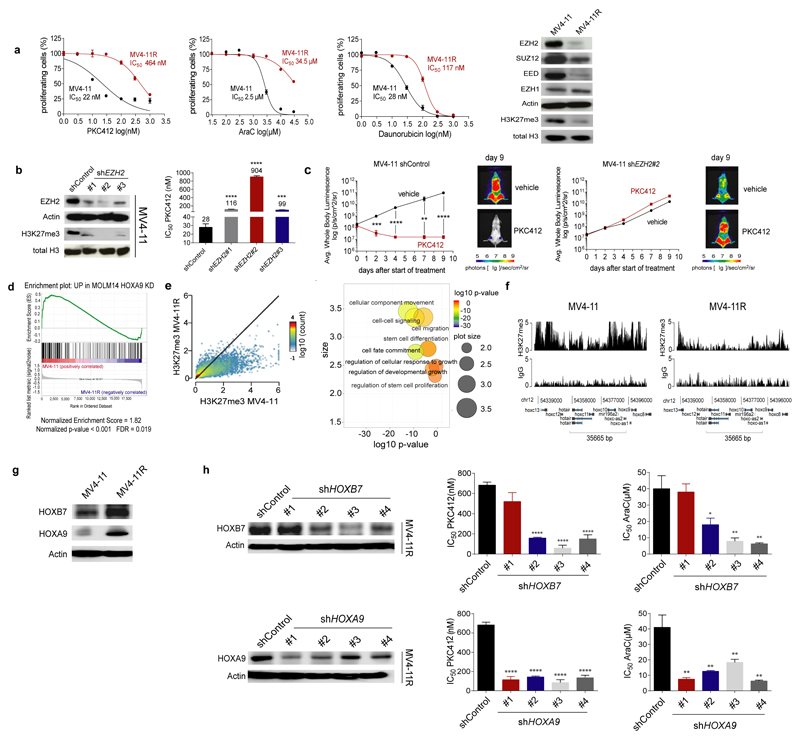
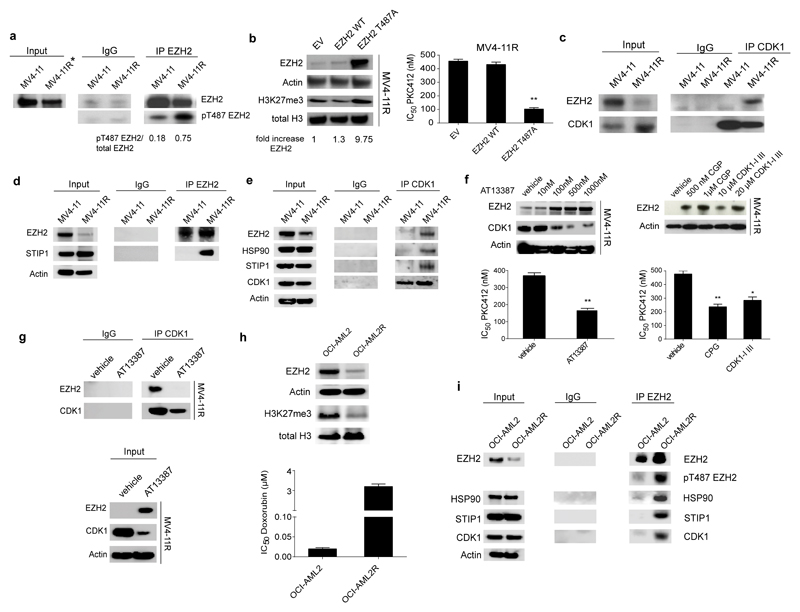
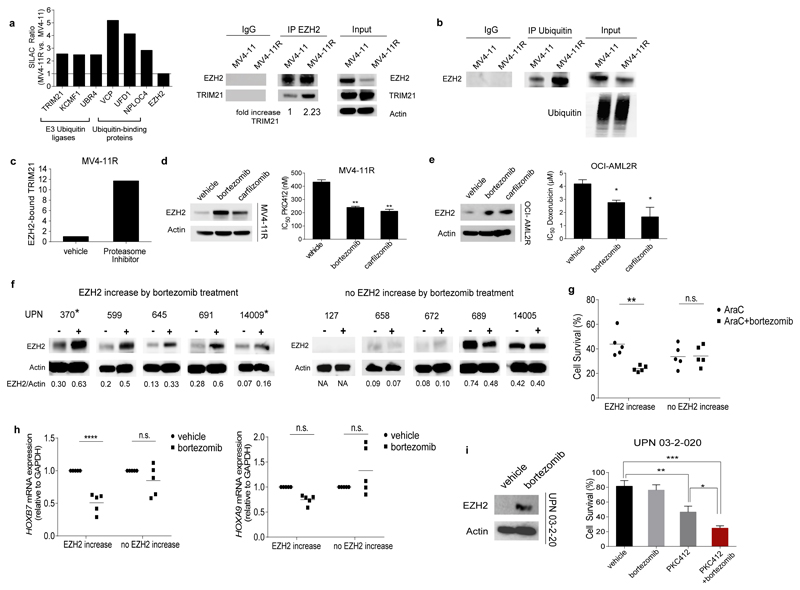
In acute myeloid leukemia (AML), therapy resistance frequently occurs, leading to high mortality among patients. However, the mechanisms that render leukemic cells drug resistant remain largely undefined. Here, we identified loss of the histone methyltransferase EZH2 and subsequent reduction of histone H3K27 trimethylation as a novel pathway of acquired resistance to tyrosine kinase inhibitors (TKIs) and cytotoxic drugs in AML. Low EZH2 protein levels correlated with poor prognosis in AML patients. Suppression of EZH2 protein expression induced chemoresistance of AML cell lines and primary cells in vitro and in vivo. Low EZH2 levels resulted in derepression of HOX genes, and knockdown of HOXB7 and HOXA9 in the resistant cells was sufficient to improve sensitivity to TKIs and cytotoxic drugs. The endogenous loss of EZH2 expression in resistant cells and primary blasts from a subset of relapsed AML patients resulted from enhanced CDK1-dependent phosphorylation of EZH2 at Thr487. This interaction was stabilized by heat shock protein 90 (HSP90) and followed by proteasomal degradation of EZH2 in drug-resistant cells. Accordingly, inhibitors of HSP90, CDK1 and the proteasome prevented EZH2 degradation, decreased HOX gene expression and restored drug sensitivity. Finally, patients with reduced EZH2 levels at progression to standard therapy responded to the combination of bortezomib and cytarabine, concomitant with the re-establishment of EZH2 expression and blast clearance. These data suggest restoration of EZH2 protein as a viable approach to overcome treatment resistance in this AML patient population.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
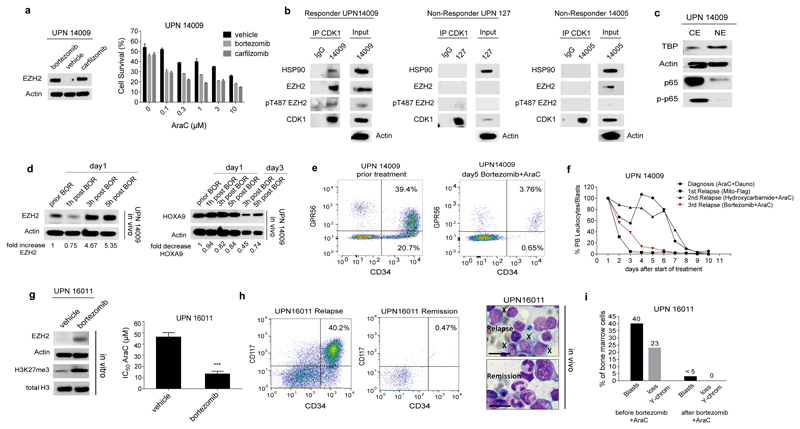
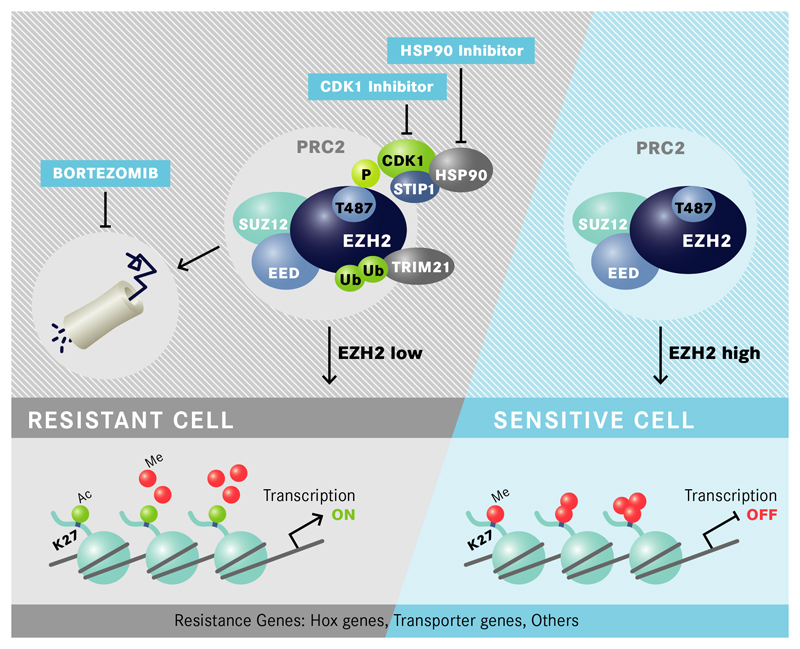
Similar articles
-
Loss-of-function mutations in the histone methyltransferase EZH2 promote chemotherapy resistance in AML.Sci Rep. 2021 Mar 12;11(1):5838. doi: 10.1038/s41598-021-84708-6. Sci Rep. 2021. PMID: 33712646 Free PMC article.
-
Nanoformulation of EPZ011989 Attenuates EZH2-c-Myb Epigenetic Interaction by Proteasomal Degradation in Acute Myeloid Leukemia.Mol Pharm. 2020 Feb 3;17(2):604-621. doi: 10.1021/acs.molpharmaceut.9b01071. Epub 2020 Jan 6. Mol Pharm. 2020. PMID: 31904978
-
Novel combination of histone methylation modulators with therapeutic synergy against acute myeloid leukemia in vitro and in vivo.Cancer Lett. 2018 Jan 28;413:35-45. doi: 10.1016/j.canlet.2017.10.015. Epub 2017 Oct 22. Cancer Lett. 2018. PMID: 29069576
-
The epigenetic role of EZH2 in acute myeloid leukemia.PeerJ. 2024 Dec 6;12:e18656. doi: 10.7717/peerj.18656. eCollection 2024. PeerJ. 2024. PMID: 39655332 Free PMC article. Review.
-
Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential.Molecules. 2024 Dec 10;29(24):5817. doi: 10.3390/molecules29245817. Molecules. 2024. PMID: 39769907 Free PMC article. Review.
Cited by
-
Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy.Cancers (Basel). 2021 Apr 6;13(7):1746. doi: 10.3390/cancers13071746. Cancers (Basel). 2021. PMID: 33917538 Free PMC article. Review.
-
Intact-Cell MALDI-ToF Mass Spectrometry for the Authentication of Drug-Adapted Cancer Cell Lines.Cells. 2019 Oct 2;8(10):1194. doi: 10.3390/cells8101194. Cells. 2019. PMID: 31581737 Free PMC article.
-
Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations-Possible Therapeutic Efficacy of Cabozantinib.Int J Mol Sci. 2019 Mar 11;20(5):1230. doi: 10.3390/ijms20051230. Int J Mol Sci. 2019. PMID: 30862120 Free PMC article.
-
TRIM21 enhances bortezomib sensitivity in multiple myeloma by halting prosurvival autophagy.Blood Adv. 2023 Oct 10;7(19):5752-5770. doi: 10.1182/bloodadvances.2022008241. Blood Adv. 2023. PMID: 37083684 Free PMC article.
-
The roles of EZH2 in cancer and its inhibitors.Med Oncol. 2023 May 6;40(6):167. doi: 10.1007/s12032-023-02025-6. Med Oncol. 2023. PMID: 37148376 Free PMC article. Review.
References
-
- Roboz GJ. Novel approaches to the treatment of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011:43–50. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
