

Complement factor H in host defense and immune evasion
- PMID: 27942748
- PMCID: PMC5378756
- DOI: 10.1007/s00018-016-2418-4
Complement factor H in host defense and immune evasion
Abstract
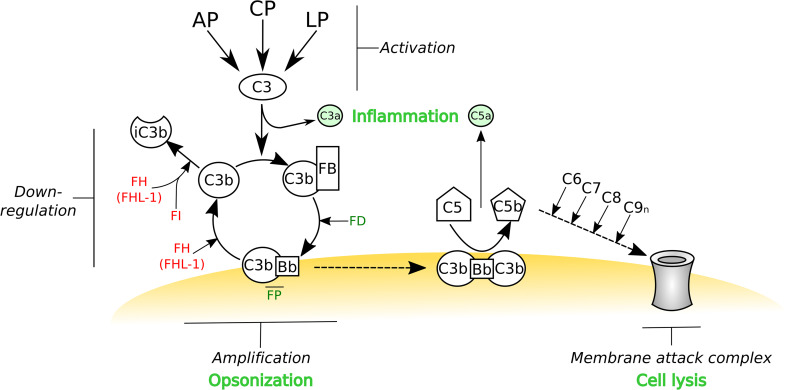
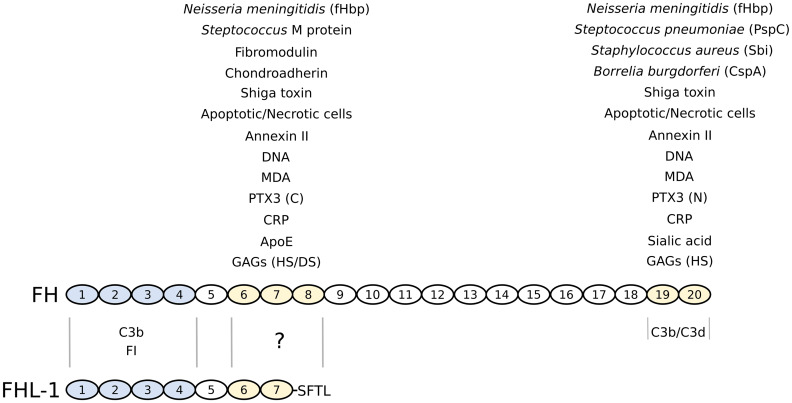
Complement is the major humoral component of the innate immune system. It recognizes pathogen- and damage-associated molecular patterns, and initiates the immune response in coordination with innate and adaptive immunity. When activated, the complement system unleashes powerful cytotoxic and inflammatory mechanisms, and thus its tight control is crucial to prevent damage to host tissues and allow restoration of immune homeostasis. Factor H is the major soluble inhibitor of complement, where its binding to self markers (i.e., particular glycan structures) prevents complement activation and amplification on host surfaces. Not surprisingly, mutations and polymorphisms that affect recognition of self by factor H are associated with diseases of complement dysregulation, such as age-related macular degeneration and atypical haemolytic uremic syndrome. In addition, pathogens (i.e., non-self) and cancer cells (i.e., altered-self) can hijack factor H to evade the immune response. Here we review recent (and not so recent) literature on the structure and function of factor H, including the emerging roles of this protein in the pathophysiology of infectious diseases and cancer.
Keywords: Cancer immunology; Complement cascade; Complement factor H; Glycan markers; Inflammatory diseases; Innate immunity.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
