

Genetic prion disease: Experience of a rapidly progressive dementia center in the United States and a review of the literature
- PMID: 27943639
- PMCID: PMC7207989
- DOI: 10.1002/ajmg.b.32505
Genetic prion disease: Experience of a rapidly progressive dementia center in the United States and a review of the literature
Abstract
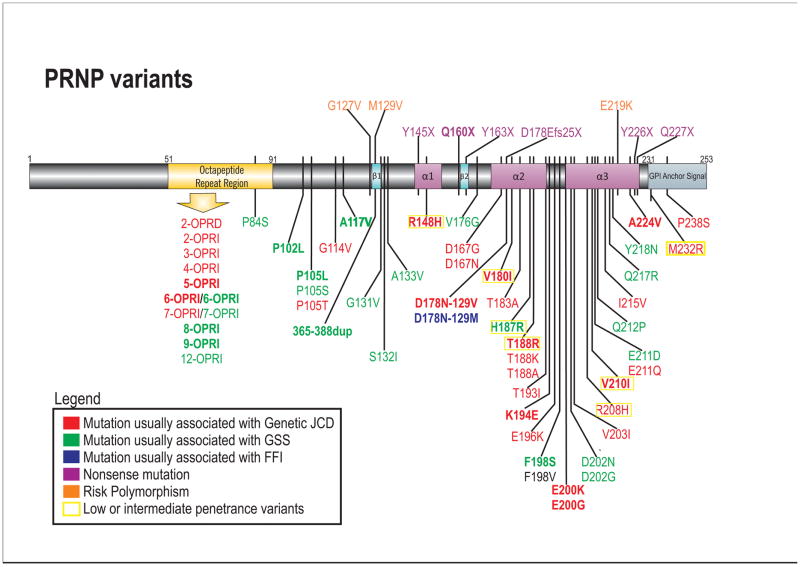
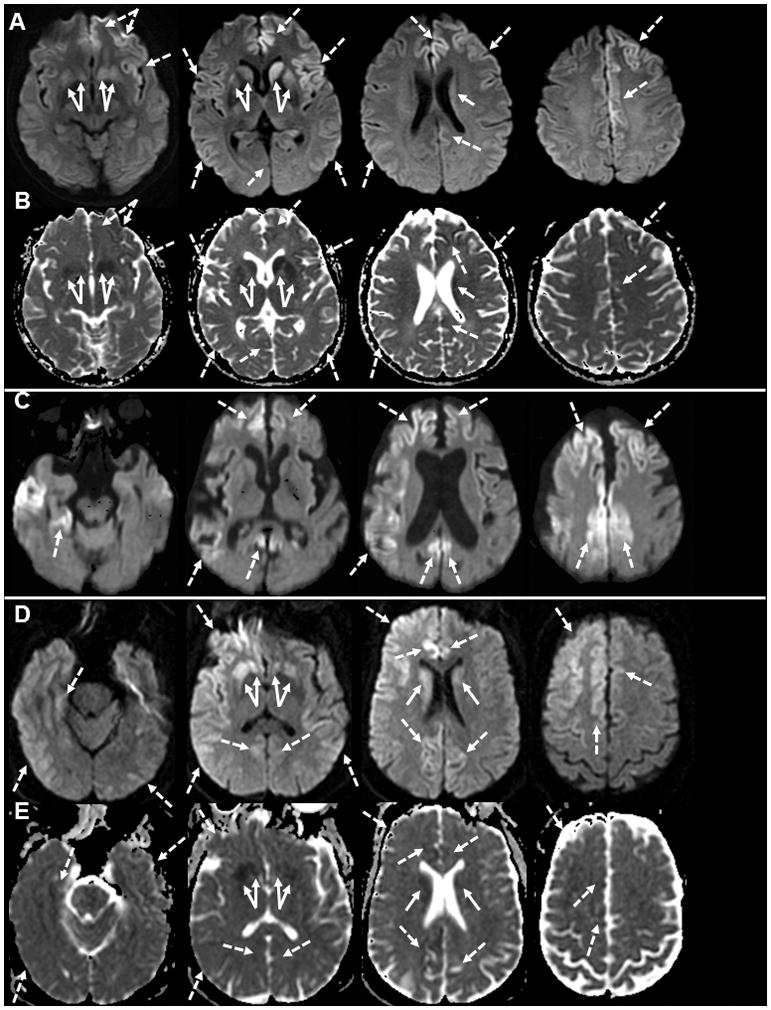
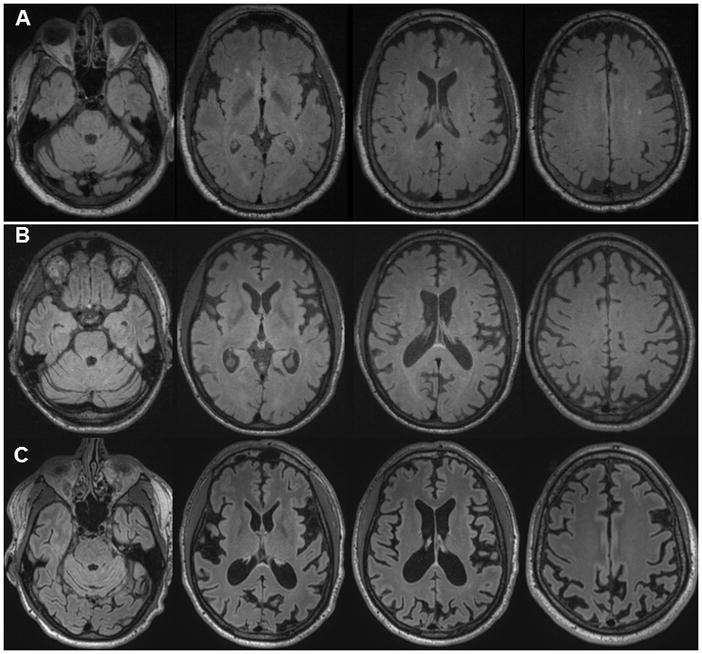
Although prion diseases are generally thought to present as rapidly progressive dementias with survival of only a few months, the phenotypic spectrum for genetic prion diseases (gPrDs) is much broader. The majority have a rapid decline with short survival, but many patients with gPrDs present as slowly progressive ataxic or parkinsonian disorders with progression over a few to several years. A few very rare mutations even present as neuropsychiatric disorders, sometimes with systemic symptoms such as gastrointestinal disorders and neuropathy, progressing over years to decades. gPrDs are caused by mutations in the prion protein gene (PRNP), and have been historically classified based on their clinicopathological features as genetic Jakob-Creutzfeldt disease (gJCD), Gerstmann-Sträussler-Scheinker (GSS), or Fatal Familial Insomnia (FFI). Mutations in PRNP can be missense, nonsense, and octapeptide repeat insertions or a deletion, and present with diverse clinical features, sensitivities of ancillary testing, and neuropathological findings. We present the UCSF gPrD cohort, including 129 symptomatic patients referred to and/or seen at UCSF between 2001 and 2016, and compare the clinical features of the gPrDs from 22 mutations identified in our cohort with data from the literature, as well as perform a literature review on most other mutations not represented in our cohort. E200K is the most common mutation worldwide, is associated with gJCD, and was the most common in the UCSF cohort. Among the GSS-associated mutations, P102L is the most commonly reported and was also the most common at UCSF. We also had several octapeptide repeat insertions (OPRI), a rare nonsense mutation (Q160X), and three novel mutations (K194E, E200G, and A224V) in our UCSF cohort. © 2016 Wiley Periodicals, Inc.
Keywords: CJD; Creutzfeldt-Jakob disease; octapeptide repeat insertion; prion protein gene; rapidly progressive dementia.
© 2016 Wiley Periodicals, Inc.
Figures
Similar articles
-
Prion disease.Handb Clin Neurol. 2018;148:441-464. doi: 10.1016/B978-0-444-64076-5.00029-6. Handb Clin Neurol. 2018. PMID: 29478593 Review.
-
Genetic PrP Prion Diseases.Cold Spring Harb Perspect Biol. 2018 May 1;10(5):a033134. doi: 10.1101/cshperspect.a033134. Cold Spring Harb Perspect Biol. 2018. PMID: 28778873 Free PMC article. Review.
-
Genetic Prion Disease: Insight from the Features and Experience of China National Surveillance for Creutzfeldt-Jakob Disease.Neurosci Bull. 2021 Nov;37(11):1570-1582. doi: 10.1007/s12264-021-00764-y. Epub 2021 Sep 6. Neurosci Bull. 2021. PMID: 34487324 Free PMC article.
-
Prion Mutations in Republic of Republic of Korea, China, and Japan.Int J Mol Sci. 2022 Dec 30;24(1):625. doi: 10.3390/ijms24010625. Int J Mol Sci. 2022. PMID: 36614069 Free PMC article. Review.
-
[Genetic background of human prion diseases].Ideggyogy Sz. 2007 Nov 30;60(11-12):438-46. Ideggyogy Sz. 2007. PMID: 18198790 Review. Hungarian.
Cited by
-
Diagnostic accuracy of cerebrospinal fluid biomarkers in genetic prion diseases.Brain. 2022 Apr 18;145(2):700-712. doi: 10.1093/brain/awab350. Brain. 2022. PMID: 35288744 Free PMC article.
-
Movement Disorders in Prionopathies: A Systematic Review.Tremor Other Hyperkinet Mov (N Y). 2019 Dec 12;9. doi: 10.7916/tohm.v0.712. eCollection 2019. Tremor Other Hyperkinet Mov (N Y). 2019. PMID: 31871824 Free PMC article.
-
A case of Gerstmann-Straussler-Scheinker (GSS) disease with supranuclear gaze palsy.J Clin Mov Disord. 2019 Dec 11;6:7. doi: 10.1186/s40734-019-0082-1. eCollection 2019. J Clin Mov Disord. 2019. PMID: 31890235 Free PMC article.
-
A case report of genetic prion disease with two different PRNP variants.Mol Genet Genomic Med. 2020 Mar;8(3):e1134. doi: 10.1002/mgg3.1134. Epub 2020 Jan 17. Mol Genet Genomic Med. 2020. PMID: 31953922 Free PMC article.
-
Early sensory disturbances and seizures are common manifestations of familial Creutzfeldt-Jakob disease due to E200K PRNP mutation: Case report from two Peruvian families.Clin Neurol Neurosurg. 2021 Mar;202:106490. doi: 10.1016/j.clineuro.2021.106490. Epub 2021 Jan 12. Clin Neurol Neurosurg. 2021. PMID: 33454496 Free PMC article. No abstract available.
References
-
- Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J, Hegyi I, Collins S, Kretzschmar H, van Duijn C, Will RG. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet. 1999;353(9165):1673–1674. - PubMed
-
- Alzualde A, Indakoetxea B, Ferrer I, Moreno F, Barandiaran M, Gorostidi A, Estanga A, Ruiz I, Calero M, van Leeuwen FW, Atares B, Juste R, Rodriguez-Martinez AB, Lopez de Munain A. A novel PRNP Y218N mutation in Gerstmann-Straussler-Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol. 2010;69(8):789–800. - PubMed
-
- Beck J, Collinge J, Mead S. Prion protein gene M232R variation is probably an uncommon polymorphism rather than a pathogenic mutation. Brain : a journal of neurology. 2012;135(Pt 2):e209. author reply e210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
