

Light Harvesting for Organic Photovoltaics
- PMID: 27951633
- PMCID: PMC5269644
- DOI: 10.1021/acs.chemrev.6b00215
Light Harvesting for Organic Photovoltaics
Abstract
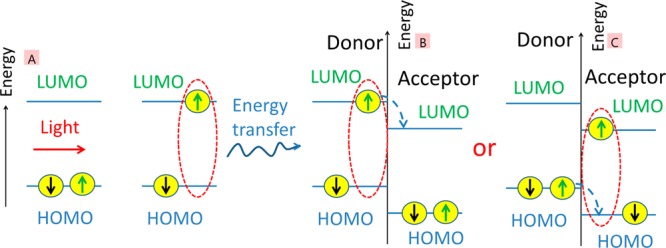
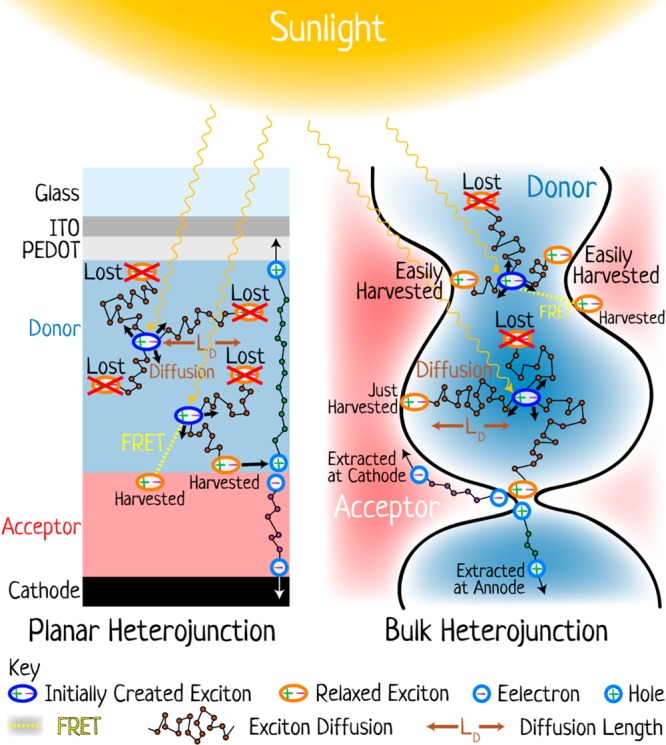
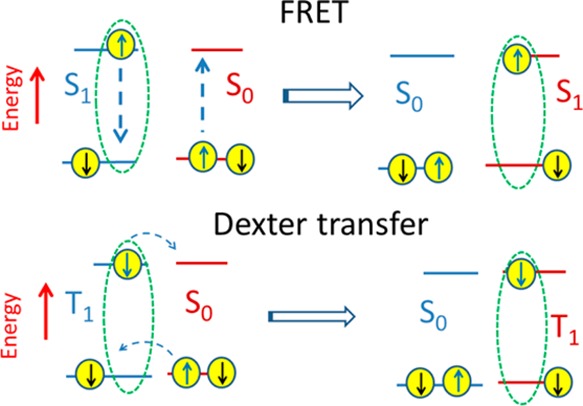
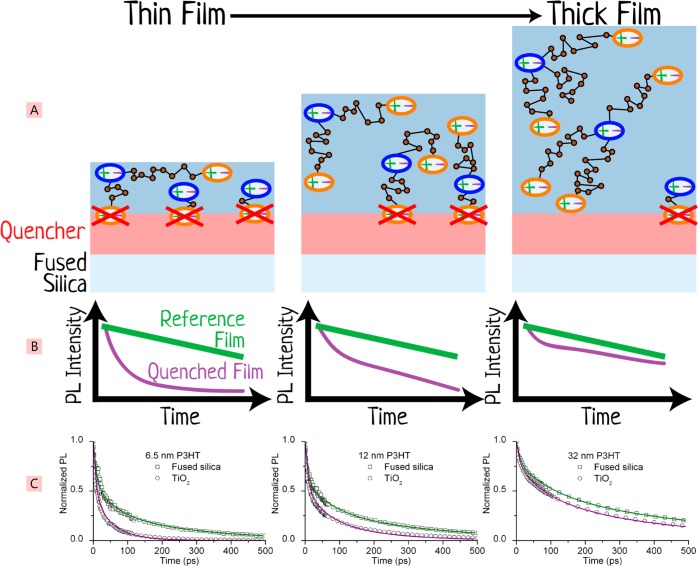
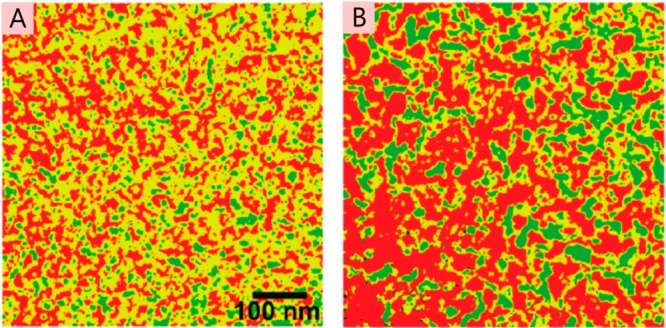
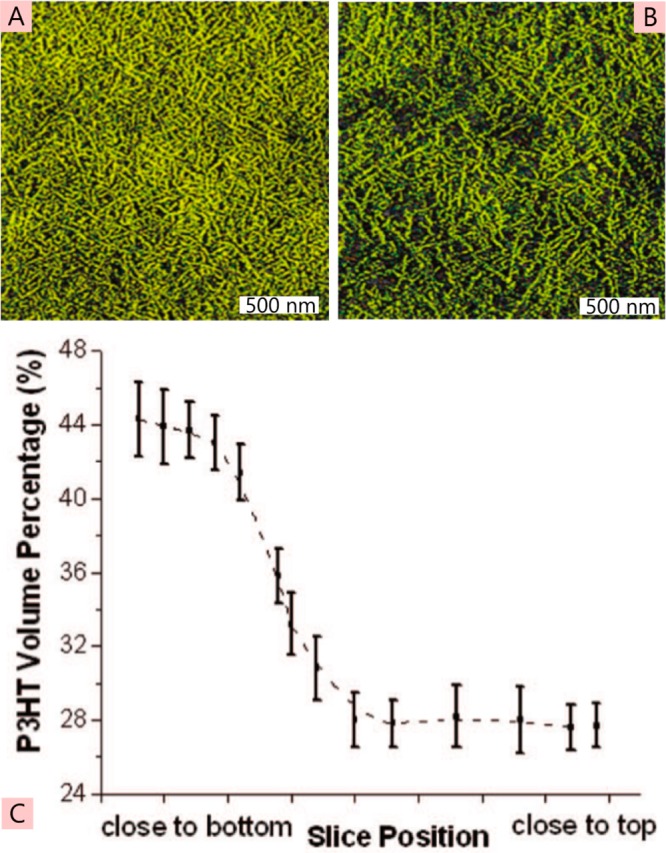
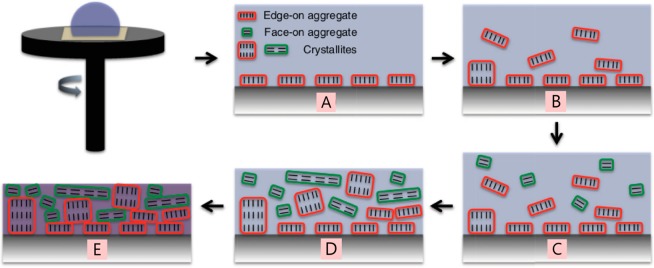
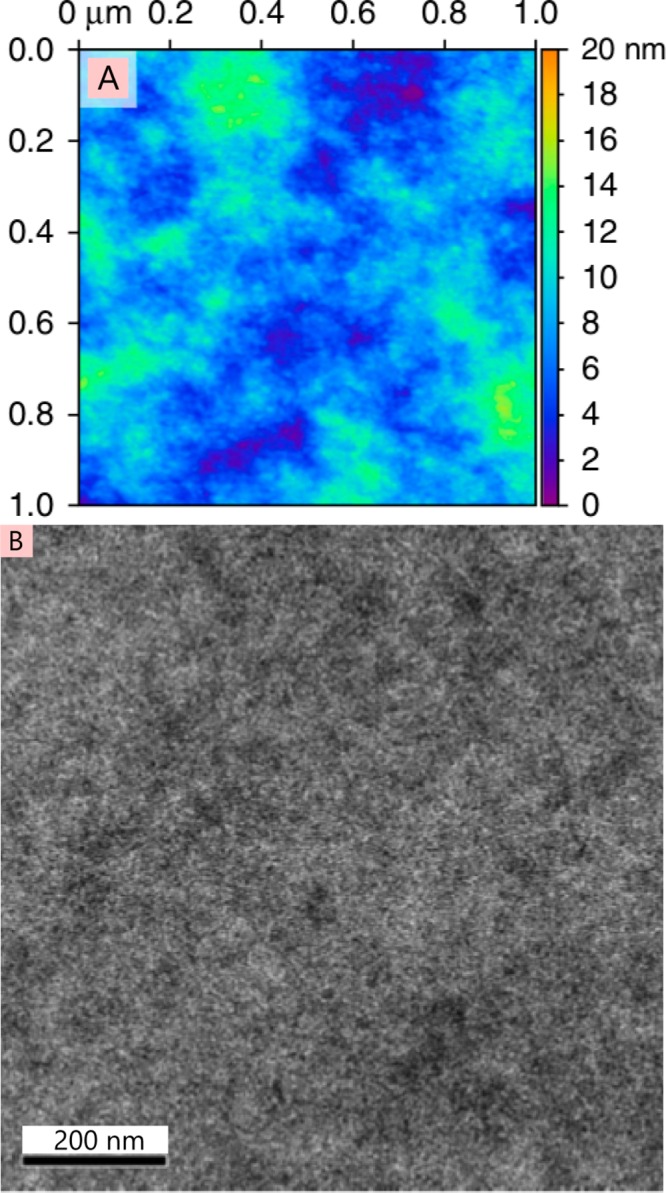
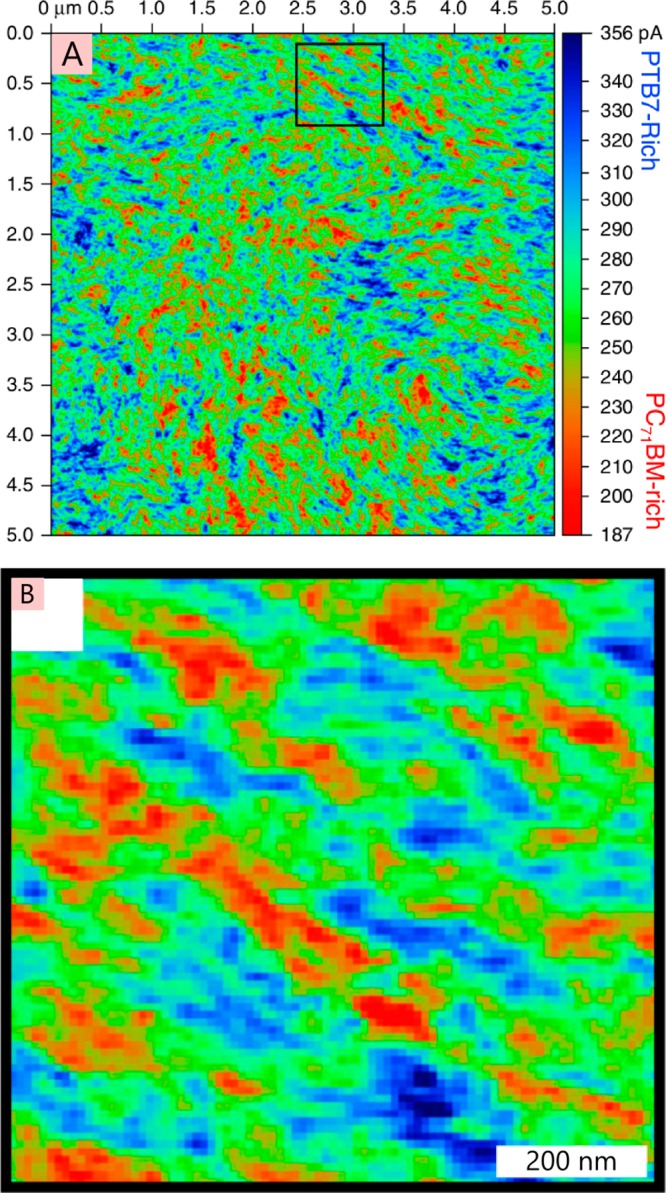
The field of organic photovoltaics has developed rapidly over the last 2 decades, and small solar cells with power conversion efficiencies of 13% have been demonstrated. Light absorbed in the organic layers forms tightly bound excitons that are split into free electrons and holes using heterojunctions of electron donor and acceptor materials, which are then extracted at electrodes to give useful electrical power. This review gives a concise description of the fundamental processes in photovoltaic devices, with the main emphasis on the characterization of energy transfer and its role in dictating device architecture, including multilayer planar heterojunctions, and on the factors that impact free carrier generation from dissociated excitons. We briefly discuss harvesting of triplet excitons, which now attracts substantial interest when used in conjunction with singlet fission. Finally, we introduce the techniques used by researchers for characterization and engineering of bulk heterojunctions to realize large photocurrents, and examine the formed morphology in three prototypical blends.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

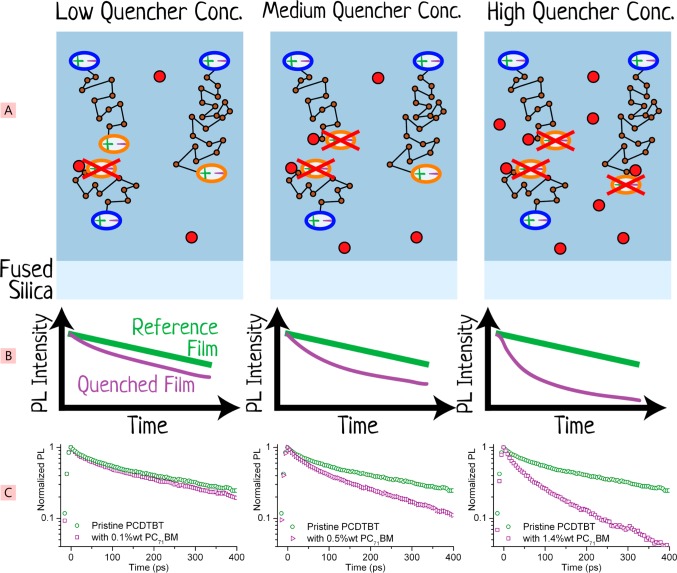

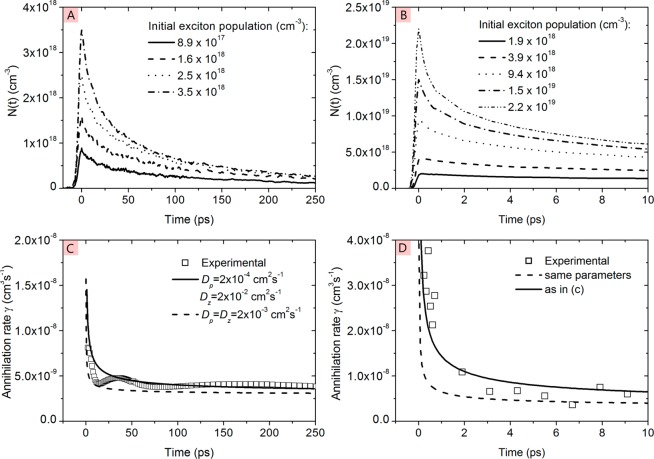
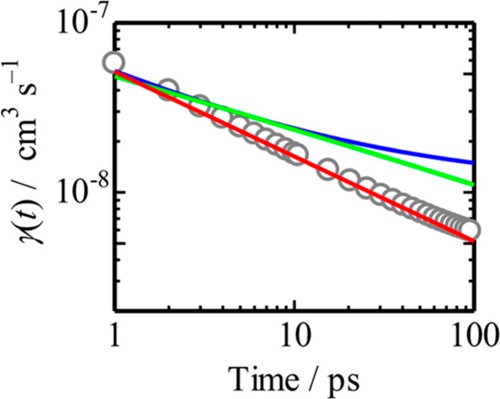







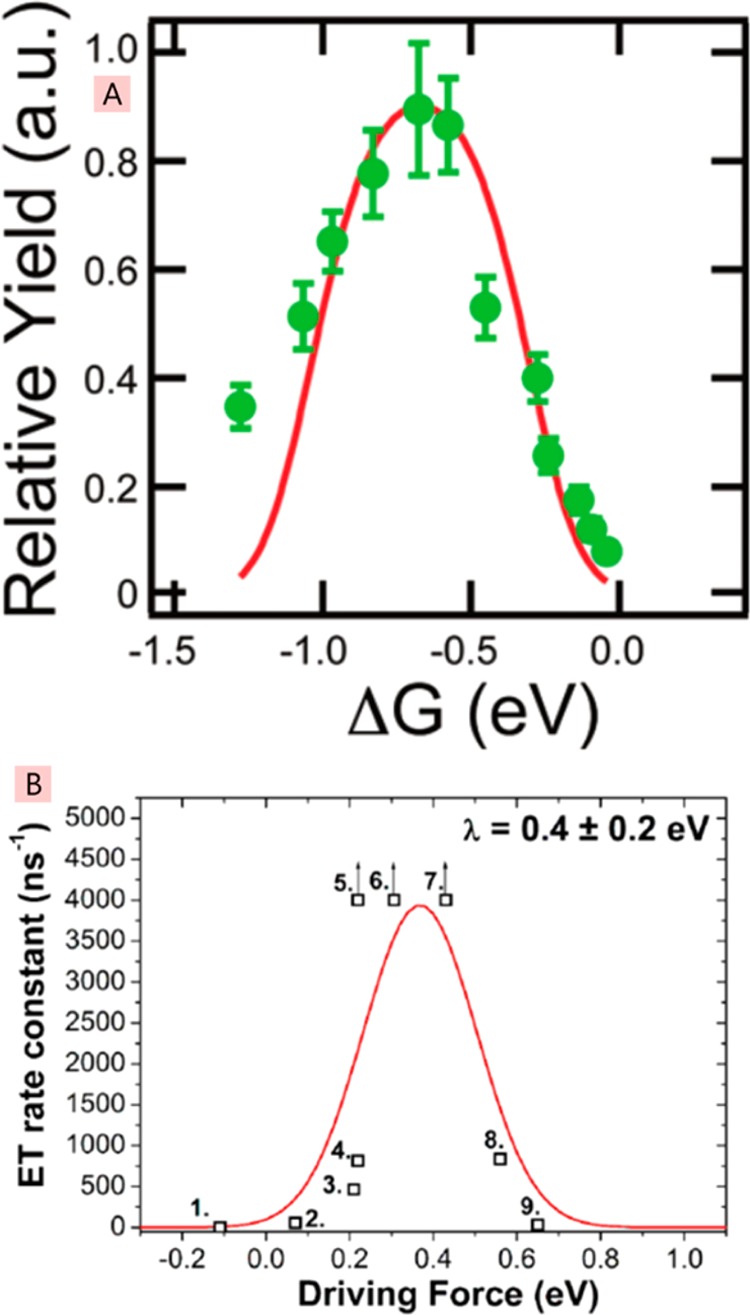
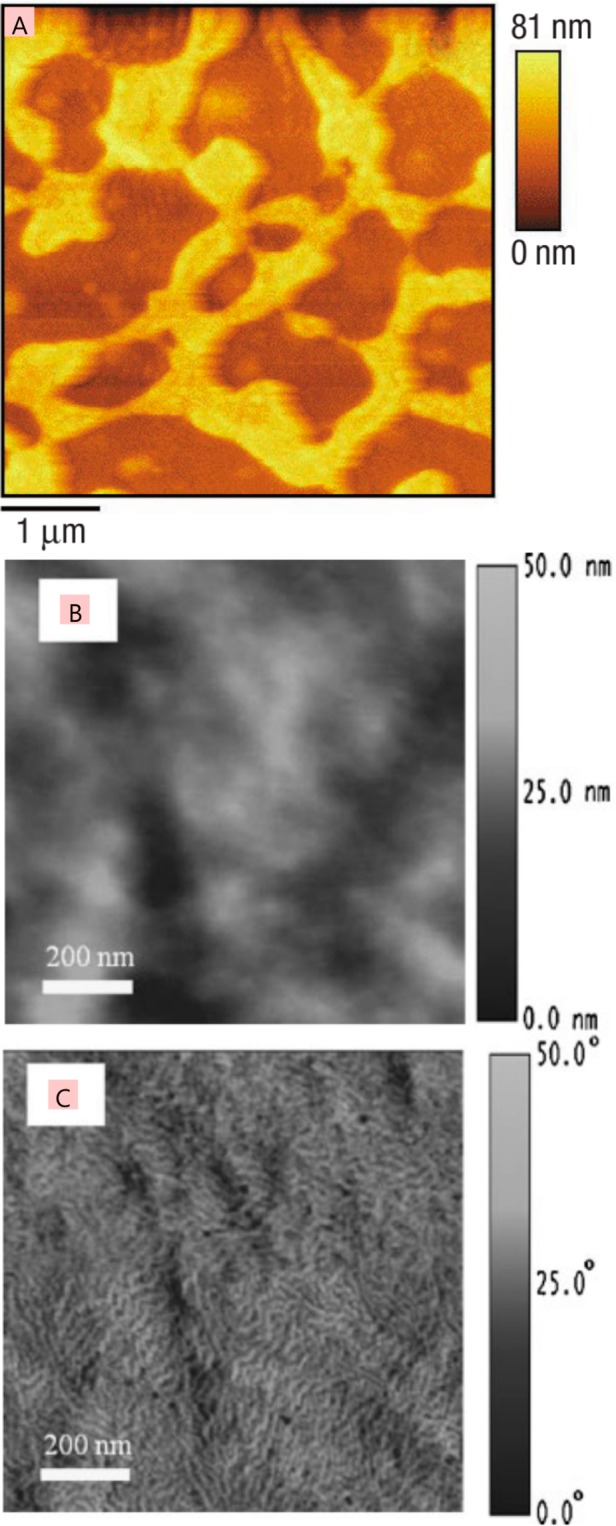
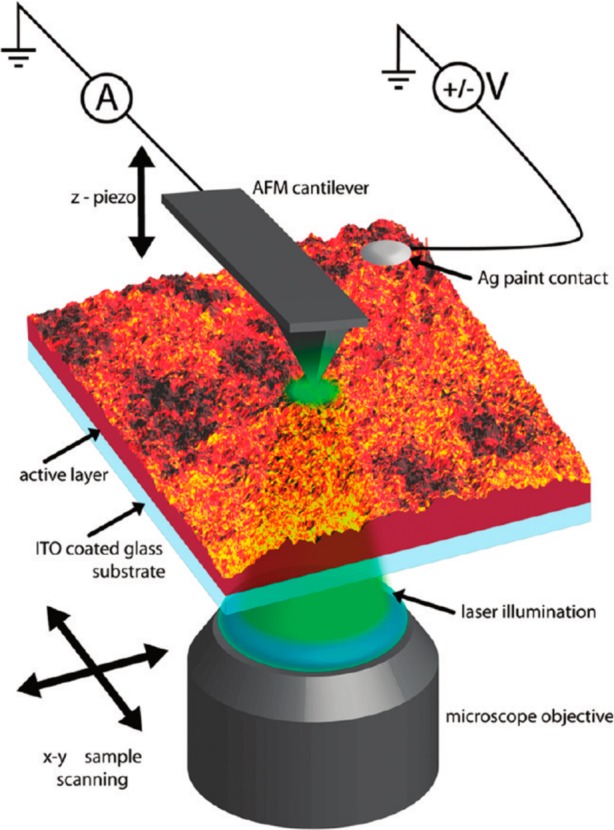




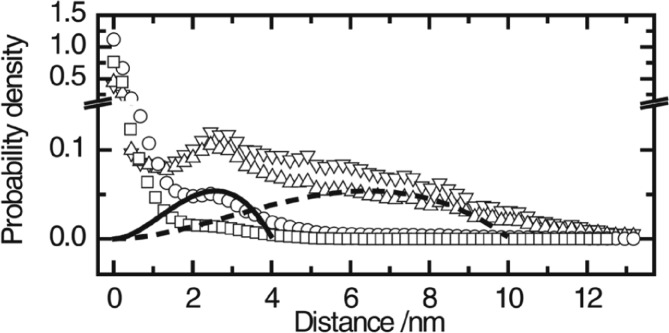




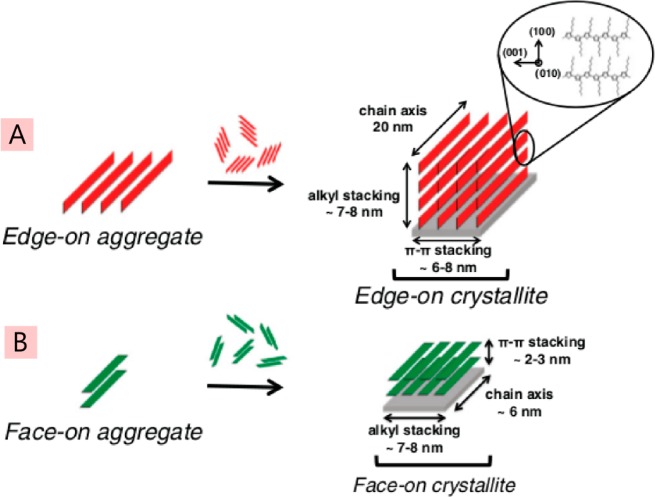








References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
