

Improved genome-scale multi-target virtual screening via a novel collaborative filtering approach to cold-start problem
- PMID: 27958331
- PMCID: PMC5153628
- DOI: 10.1038/srep38860
Improved genome-scale multi-target virtual screening via a novel collaborative filtering approach to cold-start problem
Abstract
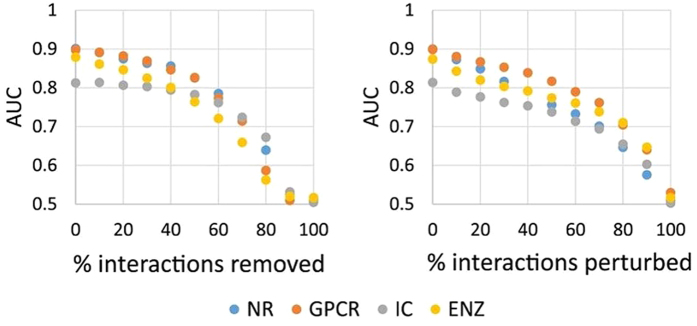
Conventional one-drug-one-gene approach has been of limited success in modern drug discovery. Polypharmacology, which focuses on searching for multi-targeted drugs to perturb disease-causing networks instead of designing selective ligands to target individual proteins, has emerged as a new drug discovery paradigm. Although many methods for single-target virtual screening have been developed to improve the efficiency of drug discovery, few of these algorithms are designed for polypharmacology. Here, we present a novel theoretical framework and a corresponding algorithm for genome-scale multi-target virtual screening based on the one-class collaborative filtering technique. Our method overcomes the sparseness of the protein-chemical interaction data by means of interaction matrix weighting and dual regularization from both chemicals and proteins. While the statistical foundation behind our method is general enough to encompass genome-wide drug off-target prediction, the program is specifically tailored to find protein targets for new chemicals with little to no available interaction data. We extensively evaluate our method using a number of the most widely accepted gene-specific and cross-gene family benchmarks and demonstrate that our method outperforms other state-of-the-art algorithms for predicting the interaction of new chemicals with multiple proteins. Thus, the proposed algorithm may provide a powerful tool for multi-target drug design.
Figures
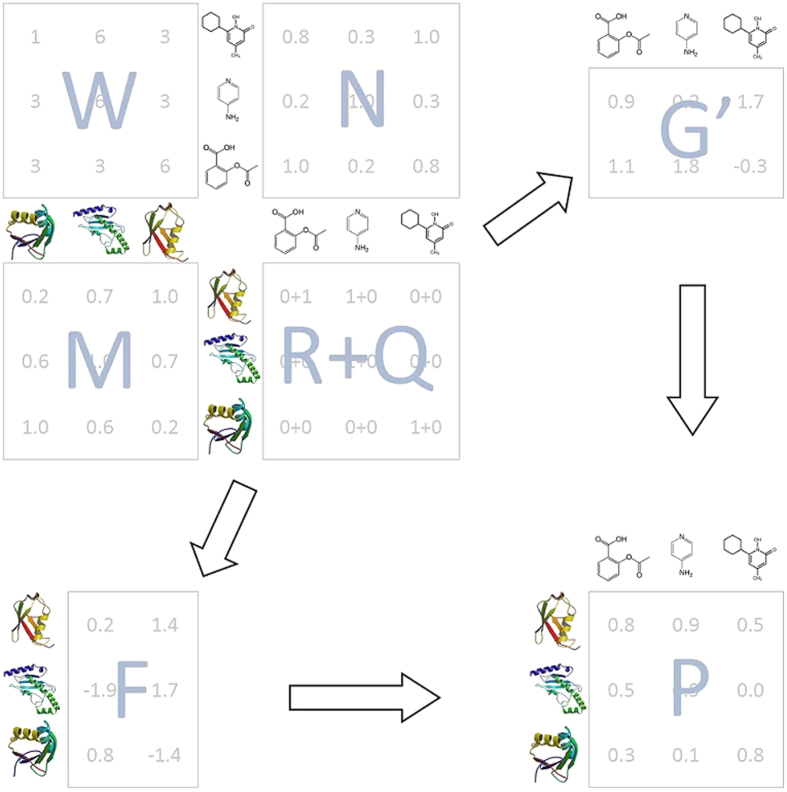
 .
.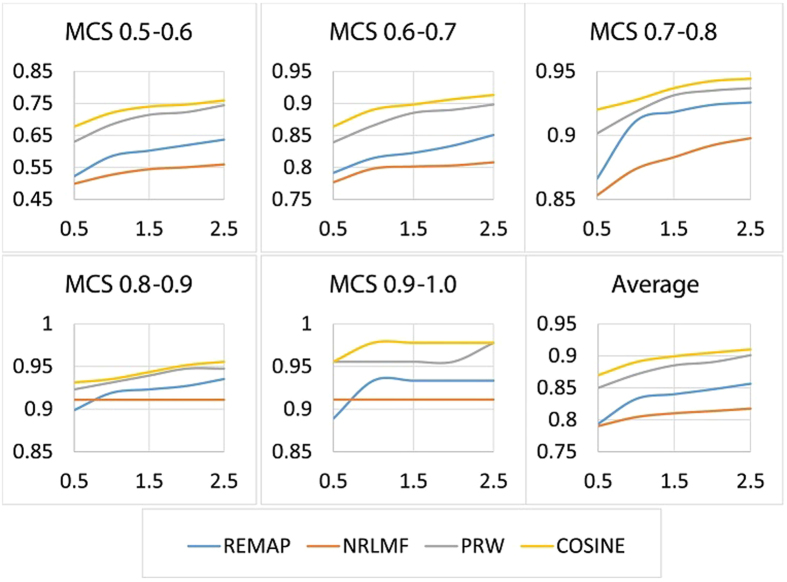
 with varying number of (maximal) chemical structural similarity (MCS).
with varying number of (maximal) chemical structural similarity (MCS).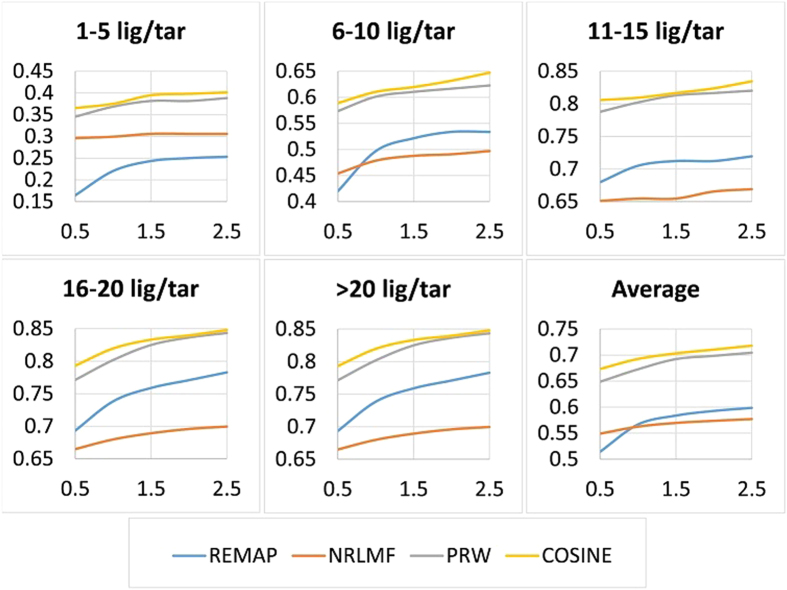
 with varying number of ligands per target (LT).
with varying number of ligands per target (LT).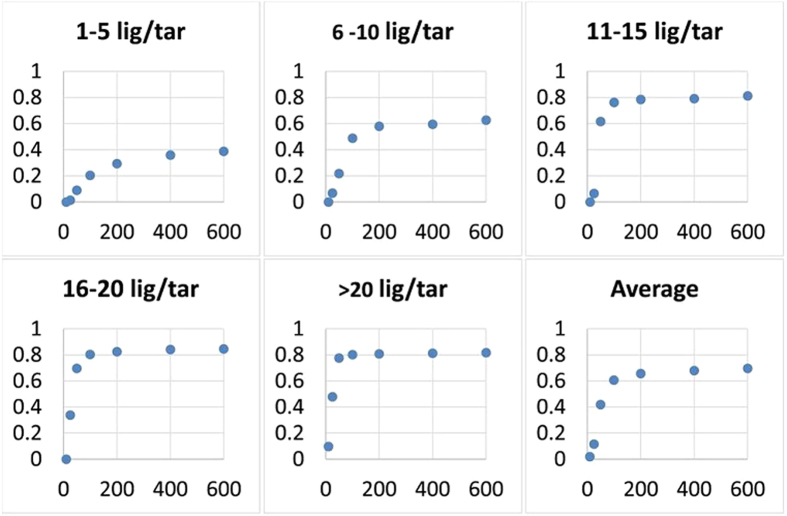
References
-
- Xie L., Xie L., Kinnings S. L. & Bourne P. E. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu. Rev. Pharmacol. Toxicol. 52, 361–379 (2012). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
