

Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation
- PMID: 27959697
- PMCID: PMC5335876
- DOI: 10.1056/NEJMoa1516767
Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation
Abstract
Background: Whole-exome sequencing can provide insight into the relationship between observed clinical phenotypes and underlying genotypes.
Methods: We conducted a retrospective analysis of data from a series of 7374 consecutive unrelated patients who had been referred to a clinical diagnostic laboratory for whole-exome sequencing; our goal was to determine the frequency and clinical characteristics of patients for whom more than one molecular diagnosis was reported. The phenotypic similarity between molecularly diagnosed pairs of diseases was calculated with the use of terms from the Human Phenotype Ontology.
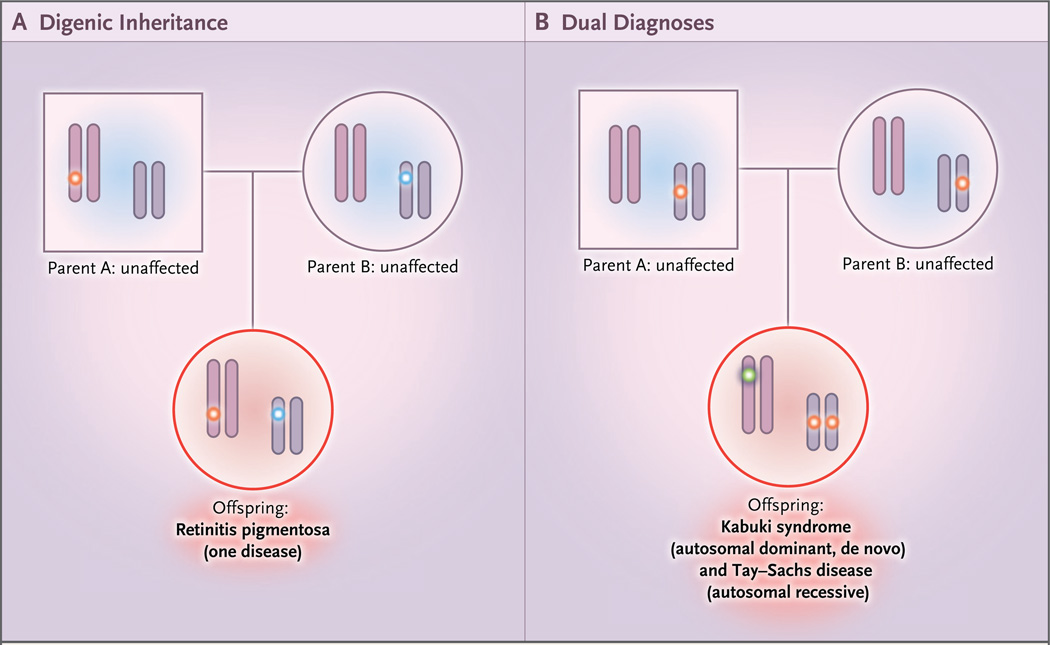
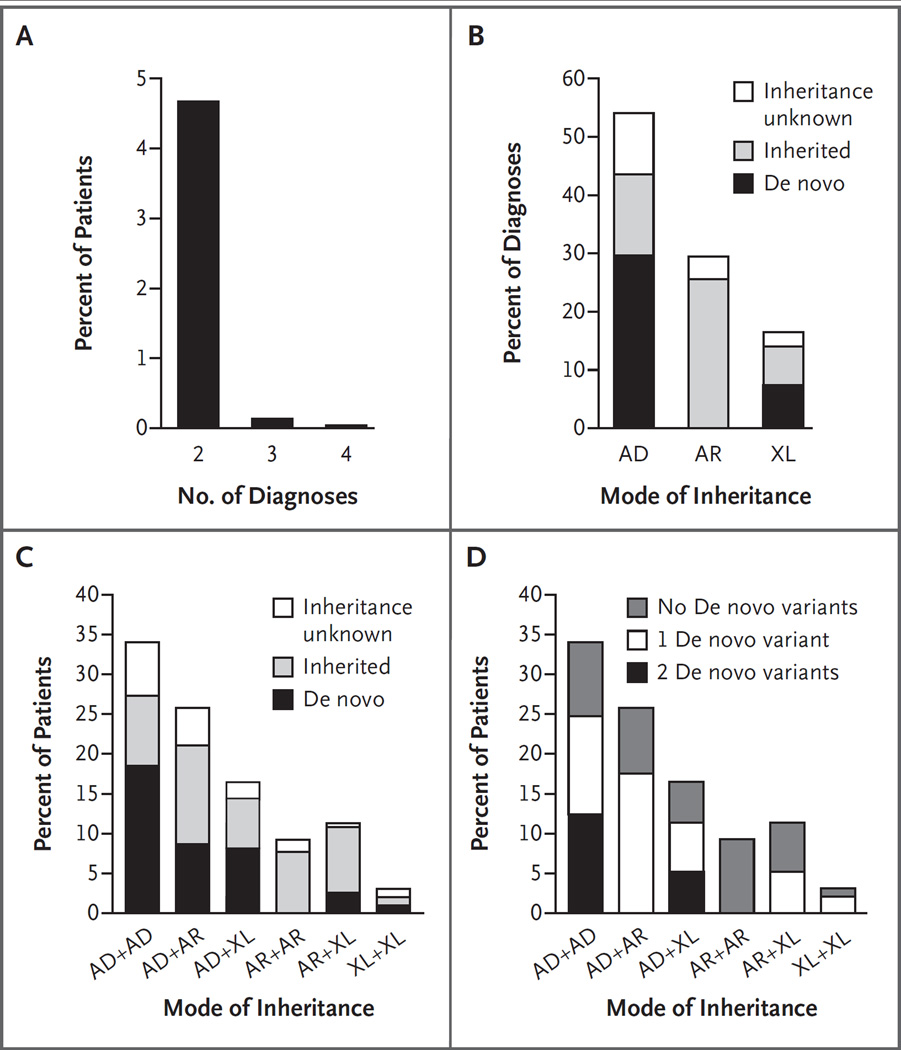
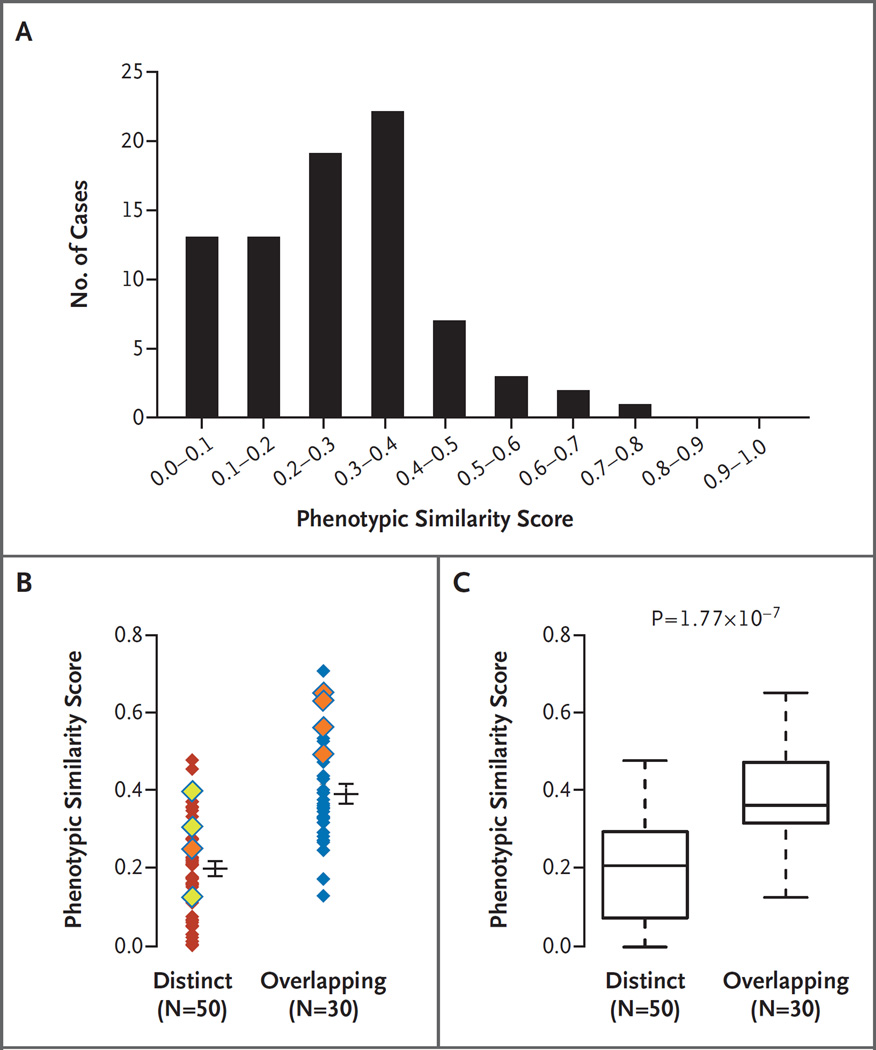
Results: A molecular diagnosis was rendered for 2076 of 7374 patients (28.2%); among these patients, 101 (4.9%) had diagnoses that involved two or more disease loci. We also analyzed parental samples, when available, and found that de novo variants accounted for 67.8% (61 of 90) of pathogenic variants in autosomal dominant disease genes and 51.7% (15 of 29) of pathogenic variants in X-linked disease genes; both variants were de novo in 44.7% (17 of 38) of patients with two monoallelic variants. Causal copy-number variants were found in 12 patients (11.9%) with multiple diagnoses. Phenotypic similarity scores were significantly lower among patients in whom the phenotype resulted from two distinct mendelian disorders that affected different organ systems (50 patients) than among patients with disorders that had overlapping phenotypic features (30 patients) (median score, 0.21 vs. 0.36; P=1.77×10-7).
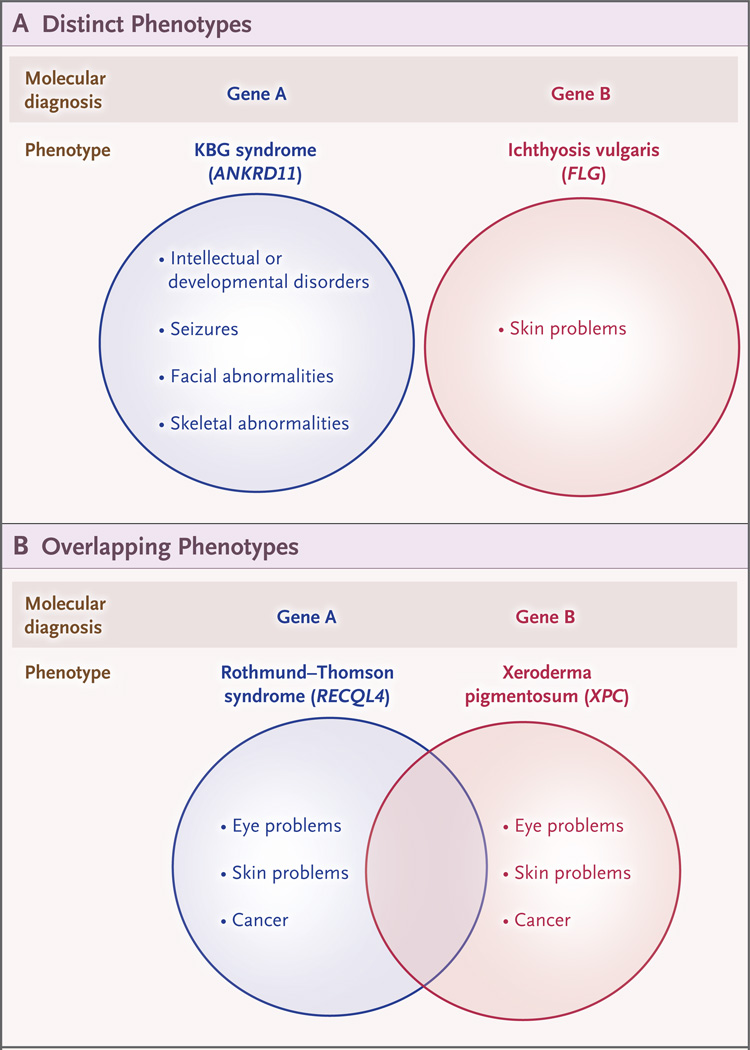
Conclusions: In our study, we found multiple molecular diagnoses in 4.9% of cases in which whole-exome sequencing was informative. Our results show that structured clinical ontologies can be used to determine the degree of overlap between two mendelian diseases in the same patient; the diseases can be distinct or overlapping. Distinct disease phenotypes affect different organ systems, whereas overlapping disease phenotypes are more likely to be caused by two genes encoding proteins that interact within the same pathway. (Funded by the National Institutes of Health and the Ting Tsung and Wei Fong Chao Foundation.).
Figures
Comment in
-
When One Diagnosis Is Not Enough.N Engl J Med. 2017 Jan 5;376(1):83-85. doi: 10.1056/NEJMe1614384. Epub 2016 Dec 7. N Engl J Med. 2017. PMID: 27959696 No abstract available.
References
-
- Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. - PubMed
-
- Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical