

Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs
- PMID: 27960273
- PMCID: PMC7717319
- DOI: 10.1021/acs.chemrev.6b00209
Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs
Abstract
Nucleoside, nucleotide, and base analogs have been in the clinic for decades to treat both viral pathogens and neoplasms. More than 20% of patients on anticancer chemotherapy have been treated with one or more of these analogs. This review focuses on the chemical synthesis and biology of anticancer nucleoside, nucleotide, and base analogs that are FDA-approved and in clinical development since 2000. We highlight the cellular biology and clinical biology of analogs, drug resistance mechanisms, and compound specificity towards different cancer types. Furthermore, we explore analog syntheses as well as improved and scale-up syntheses. We conclude with a discussion on what might lie ahead for medicinal chemists, biologists, and physicians as they try to improve analog efficacy through prodrug strategies and drug combinations.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





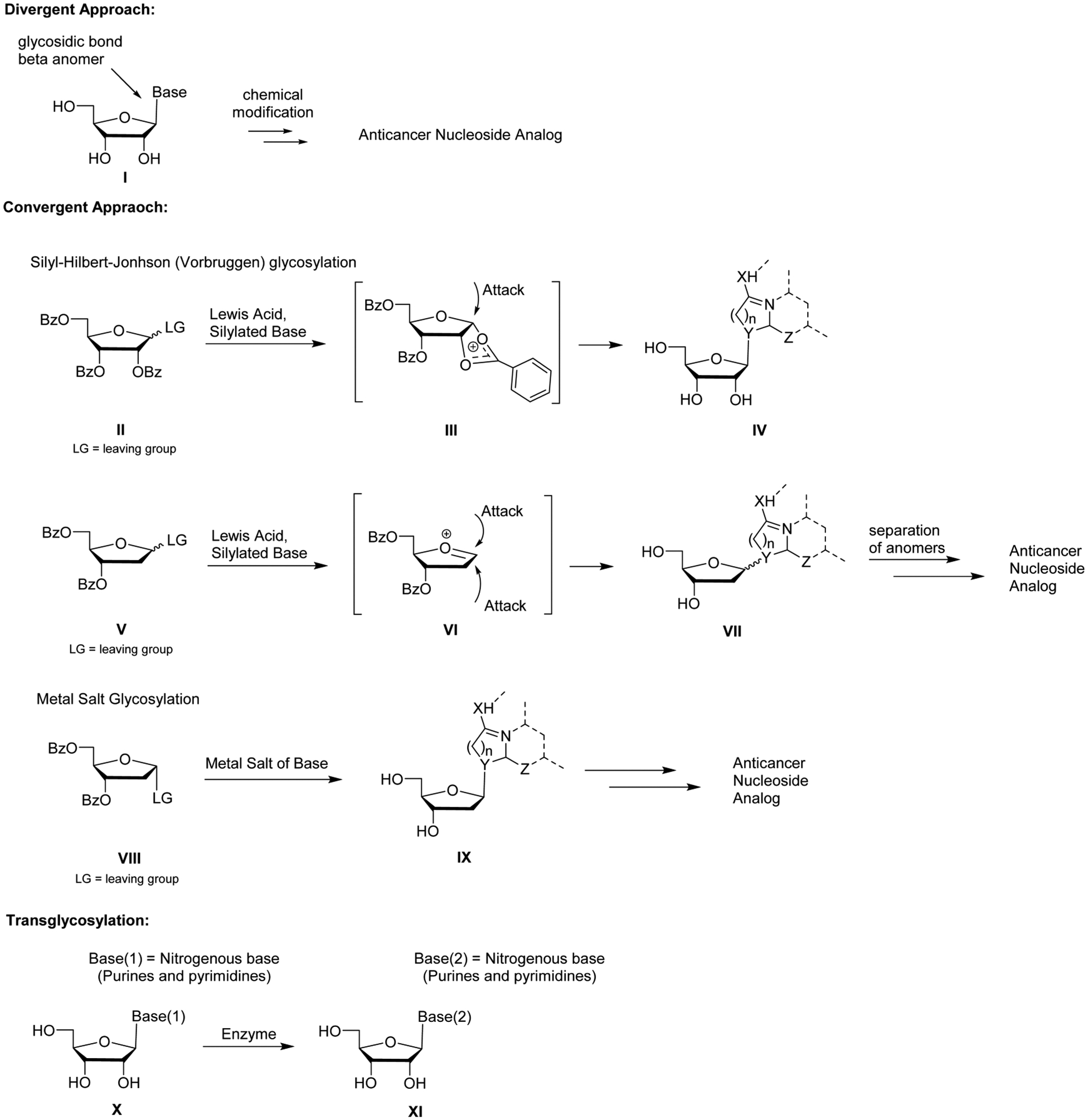

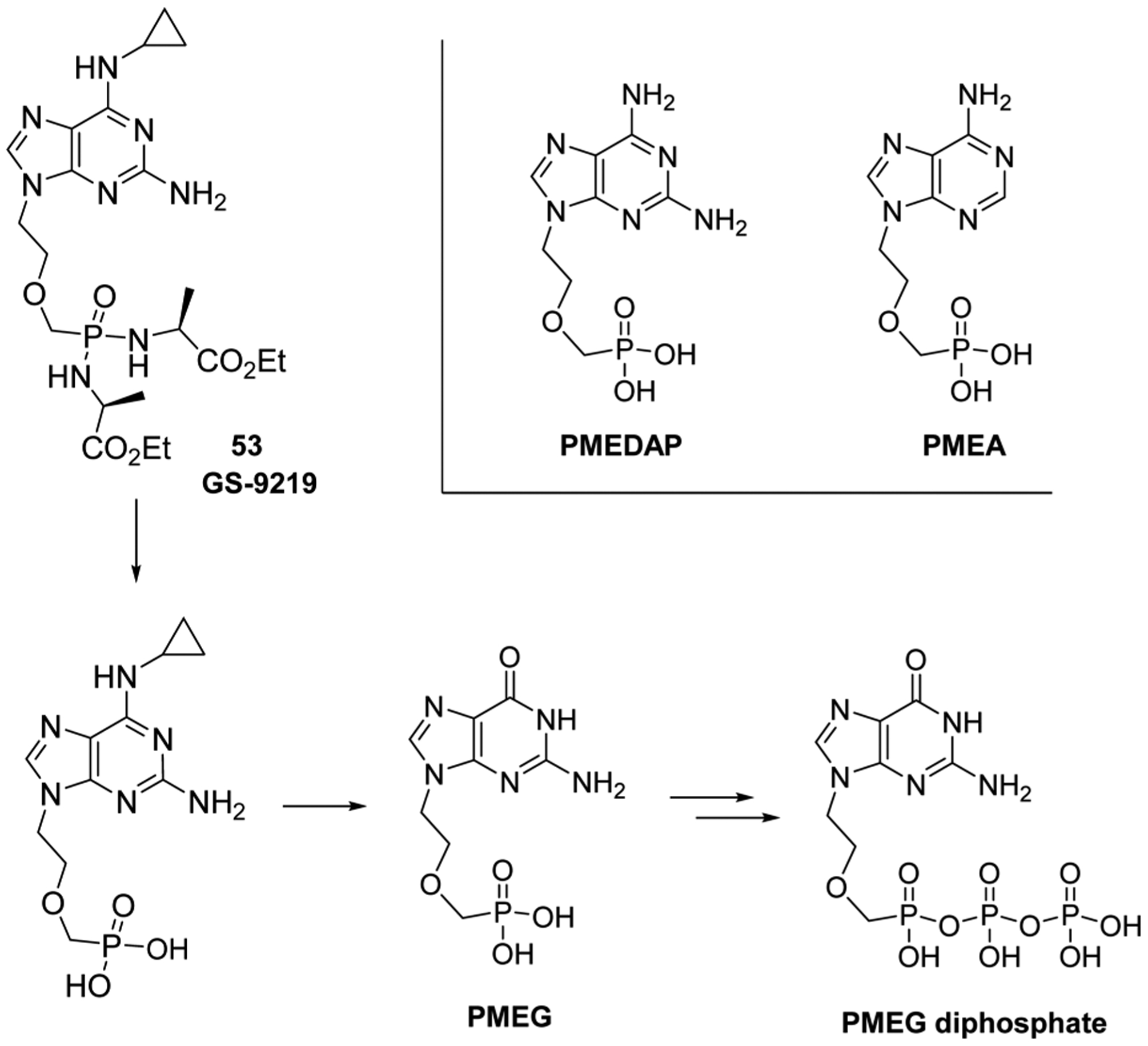

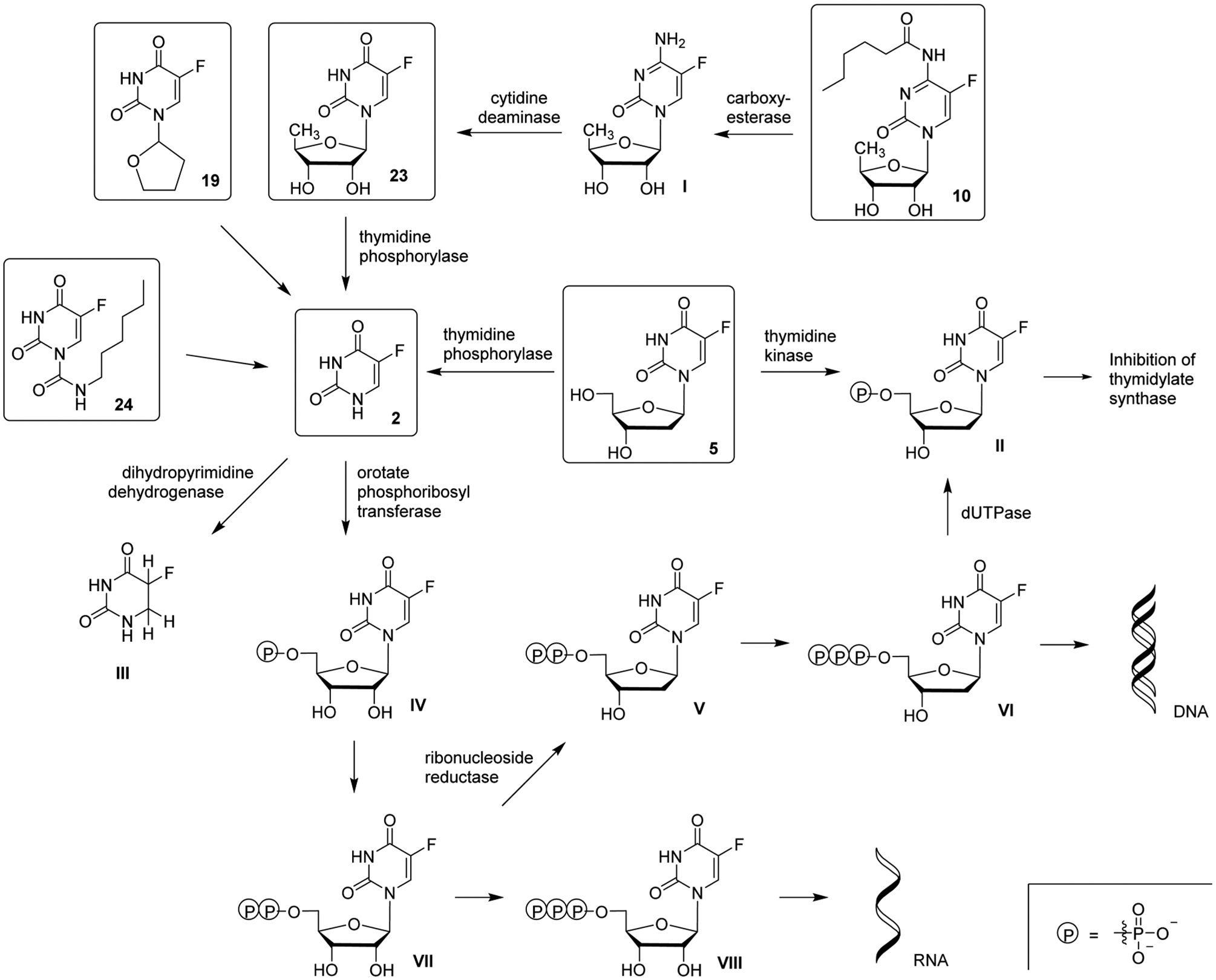



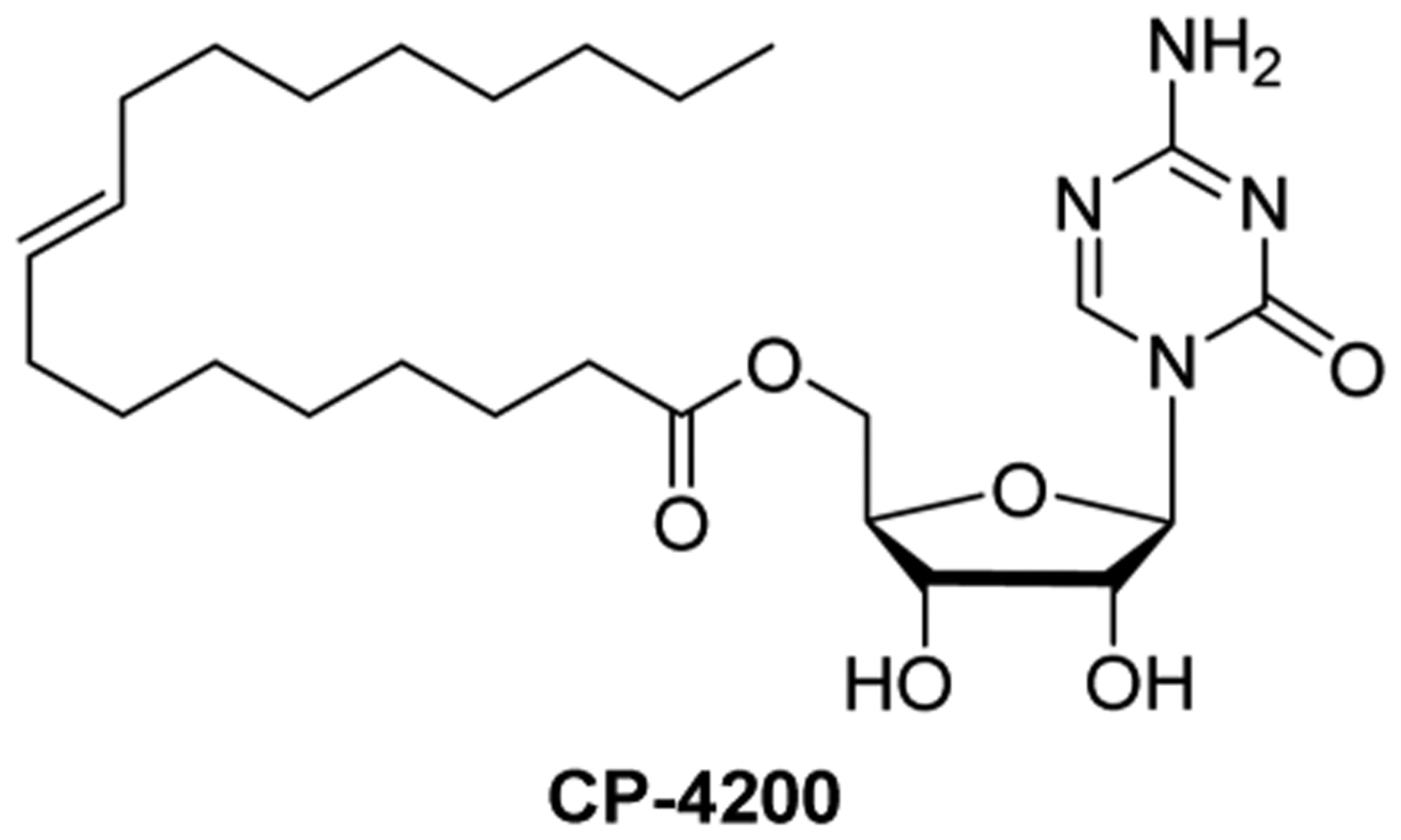
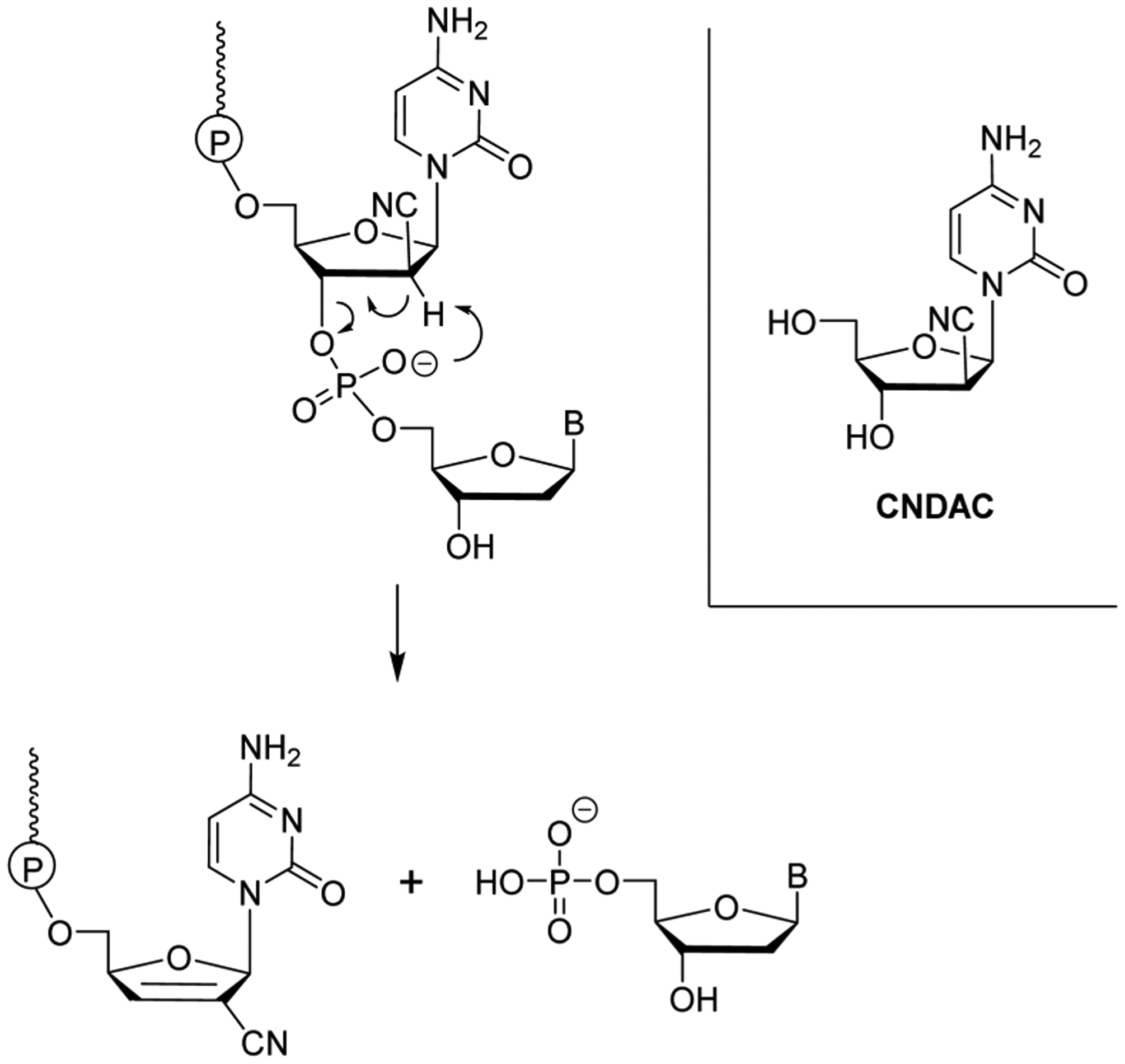



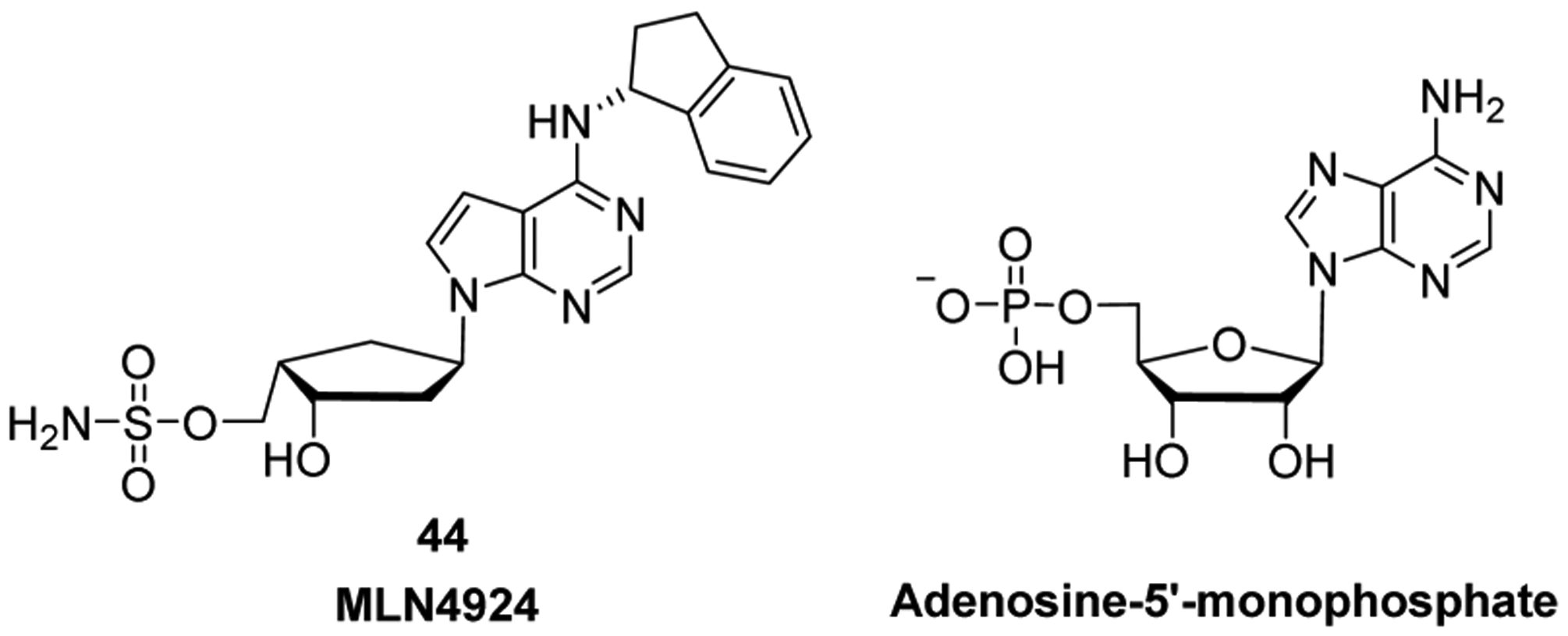


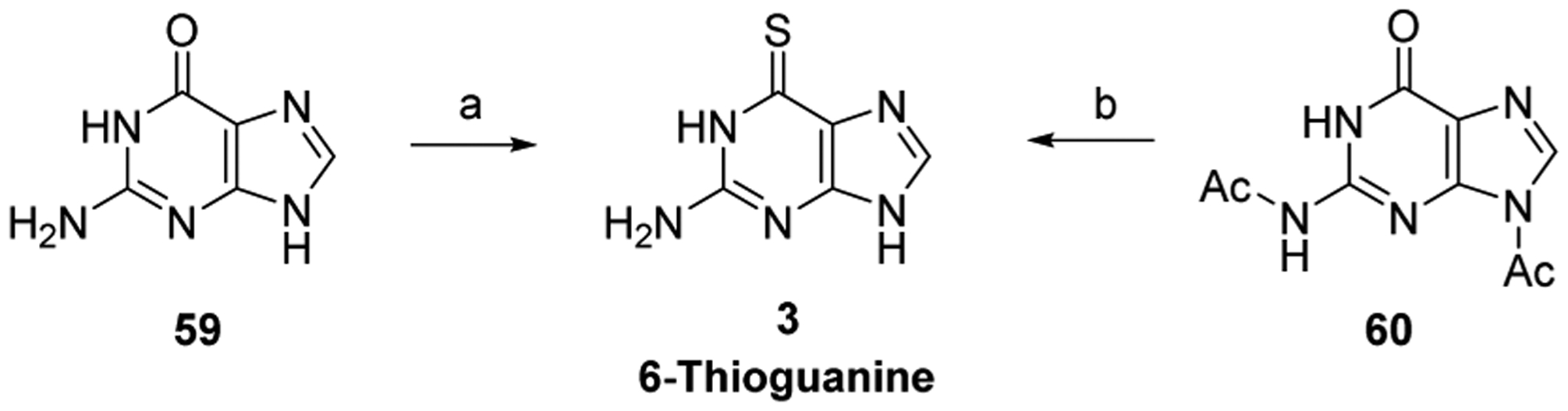

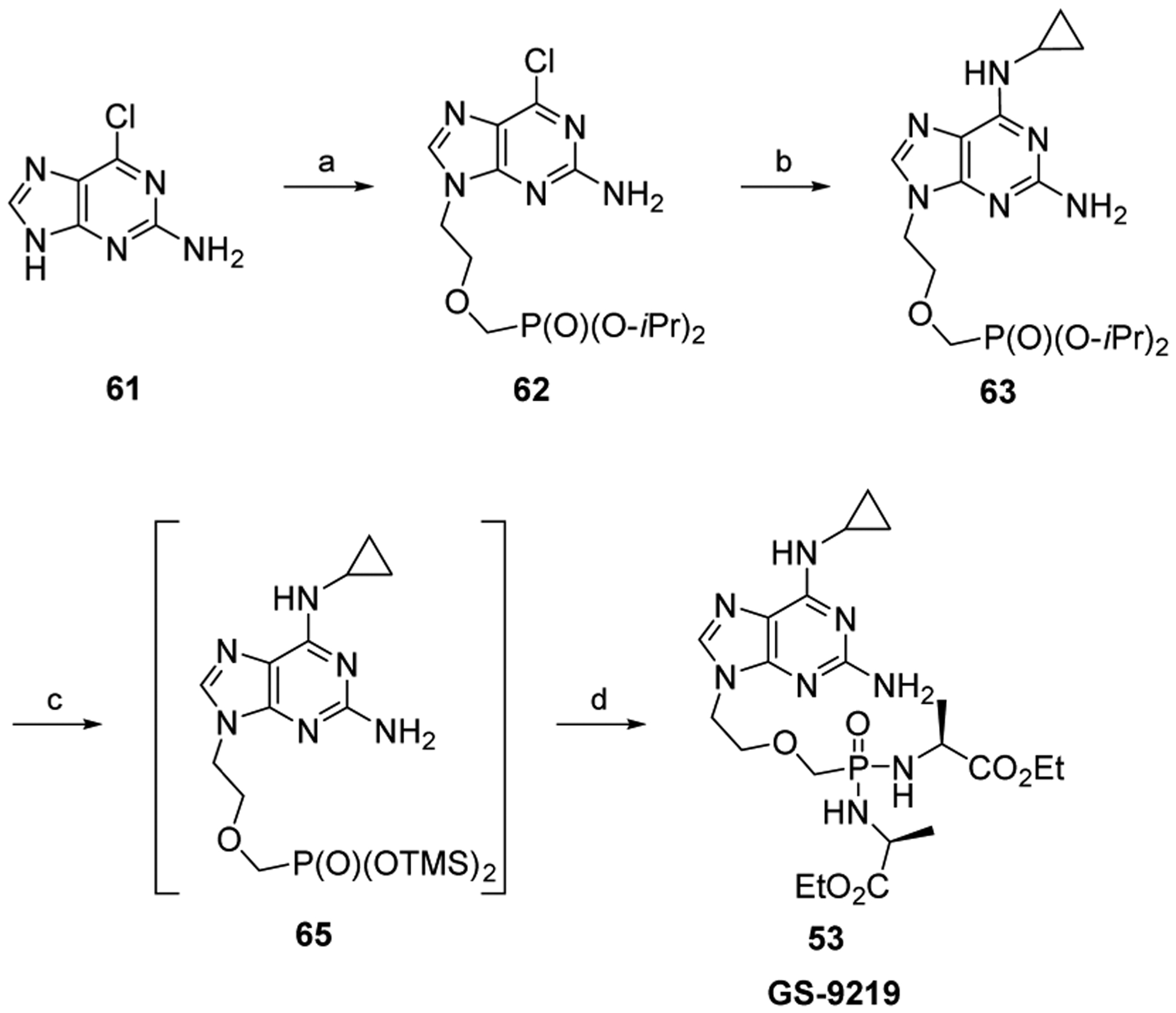





















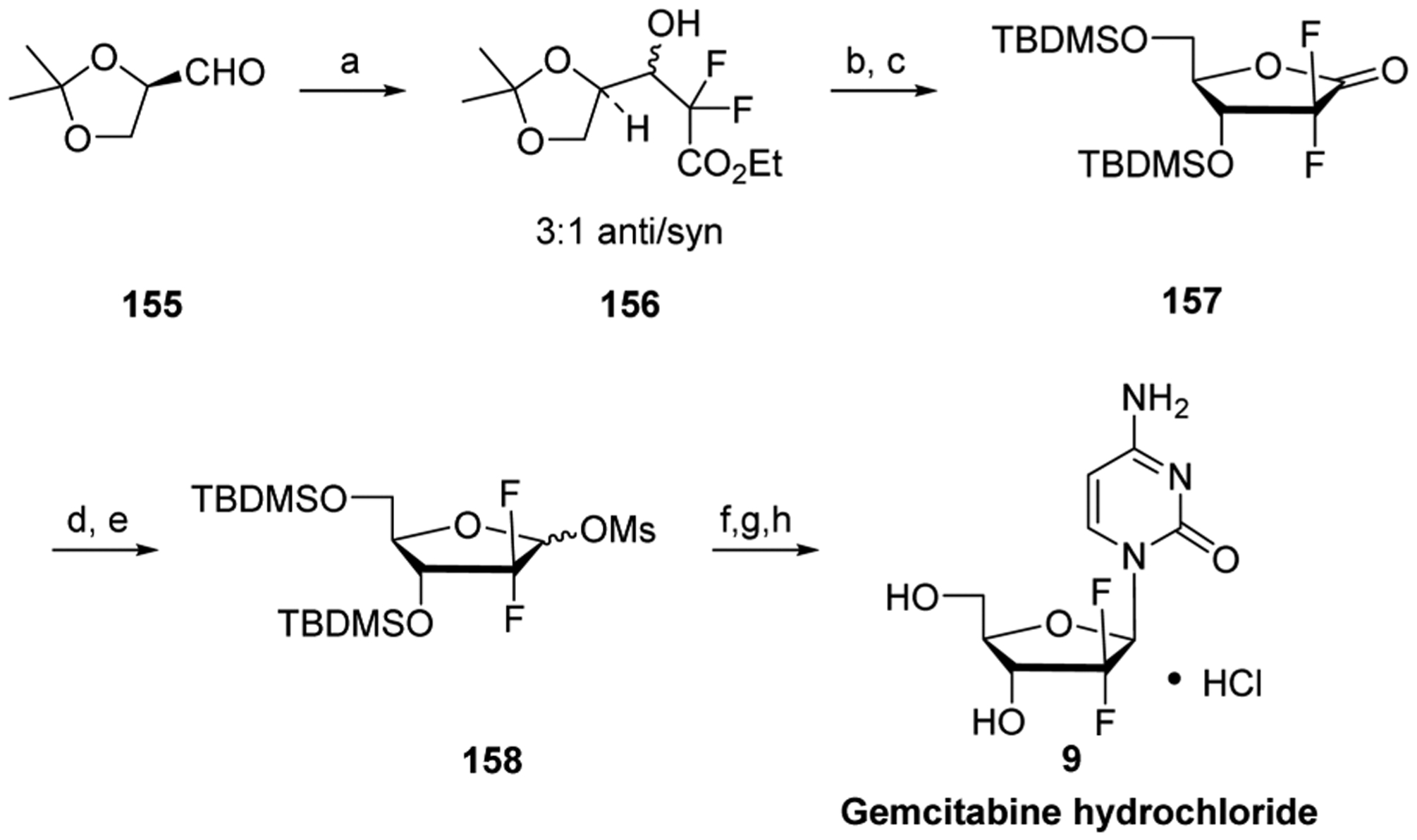


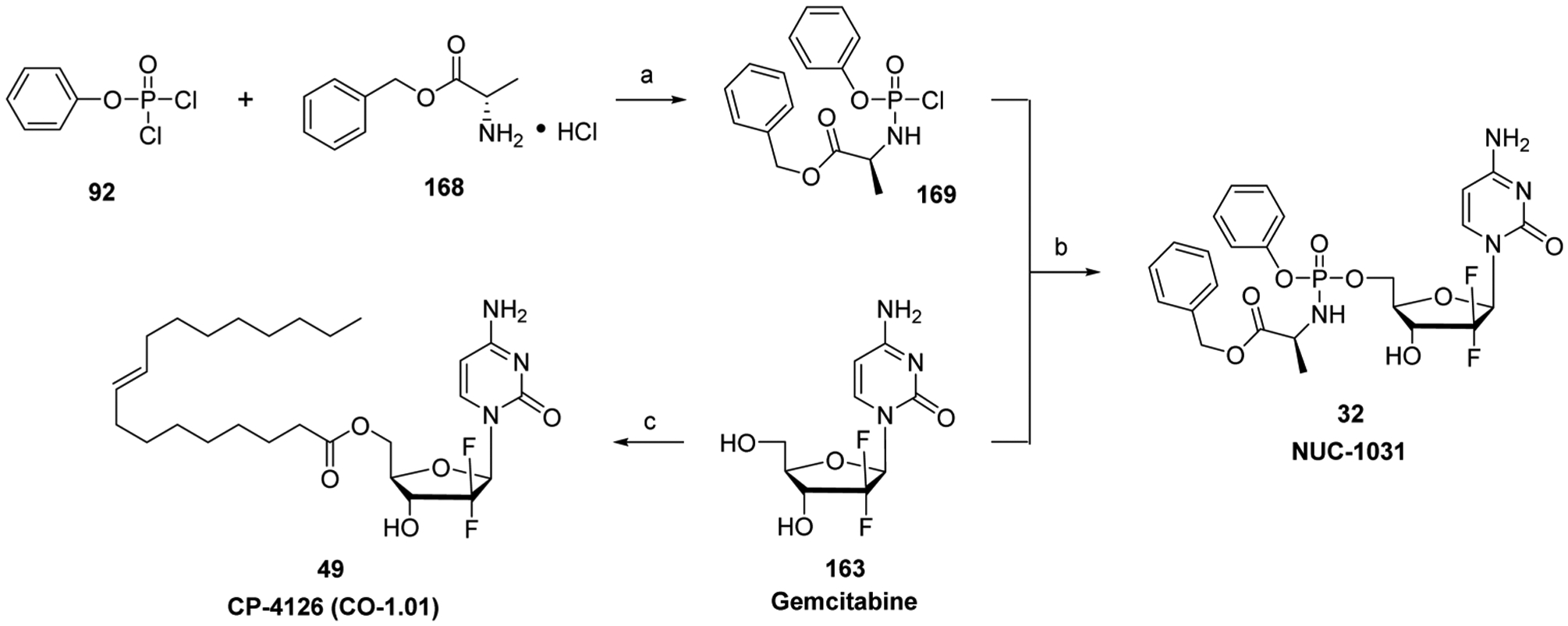

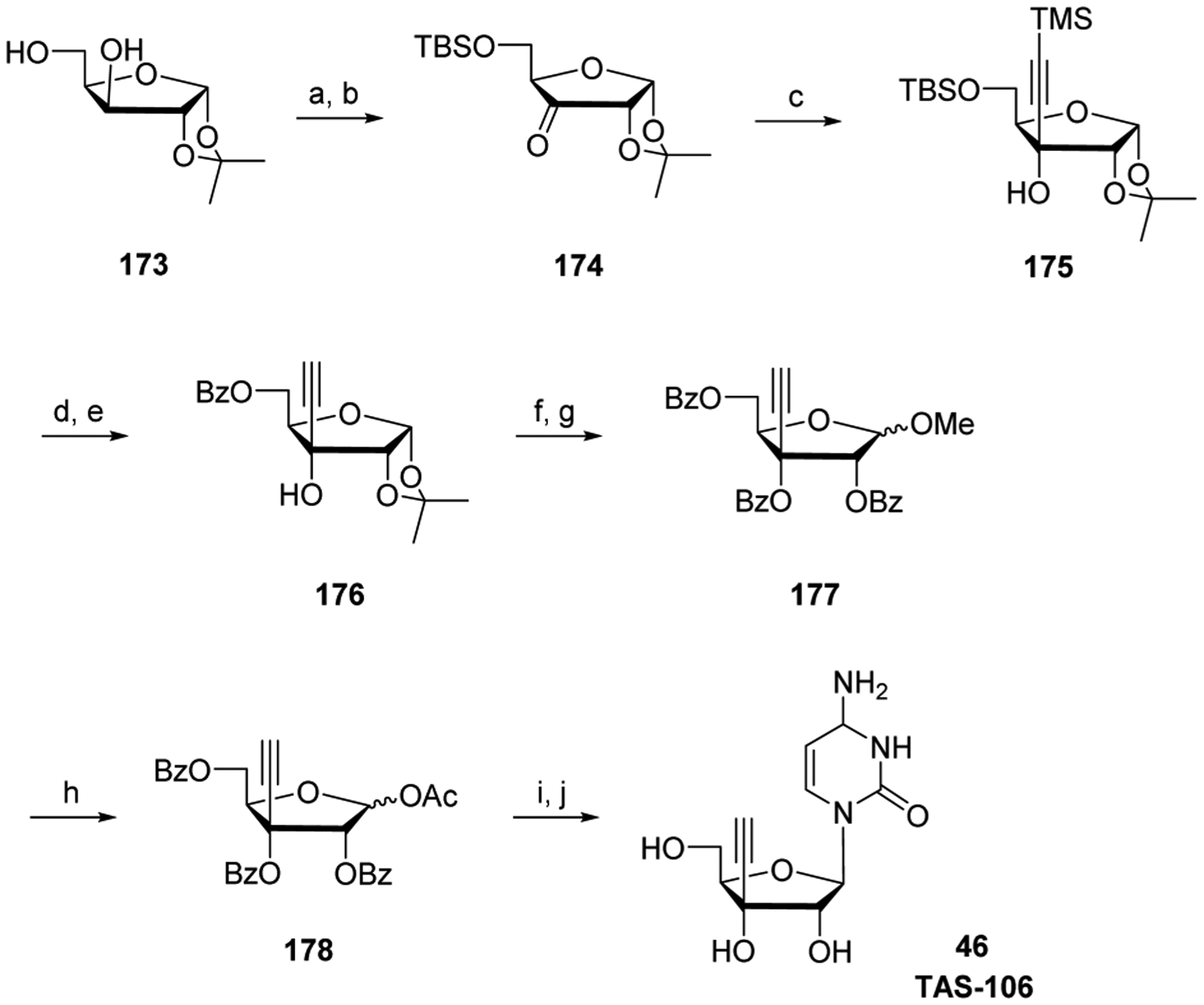








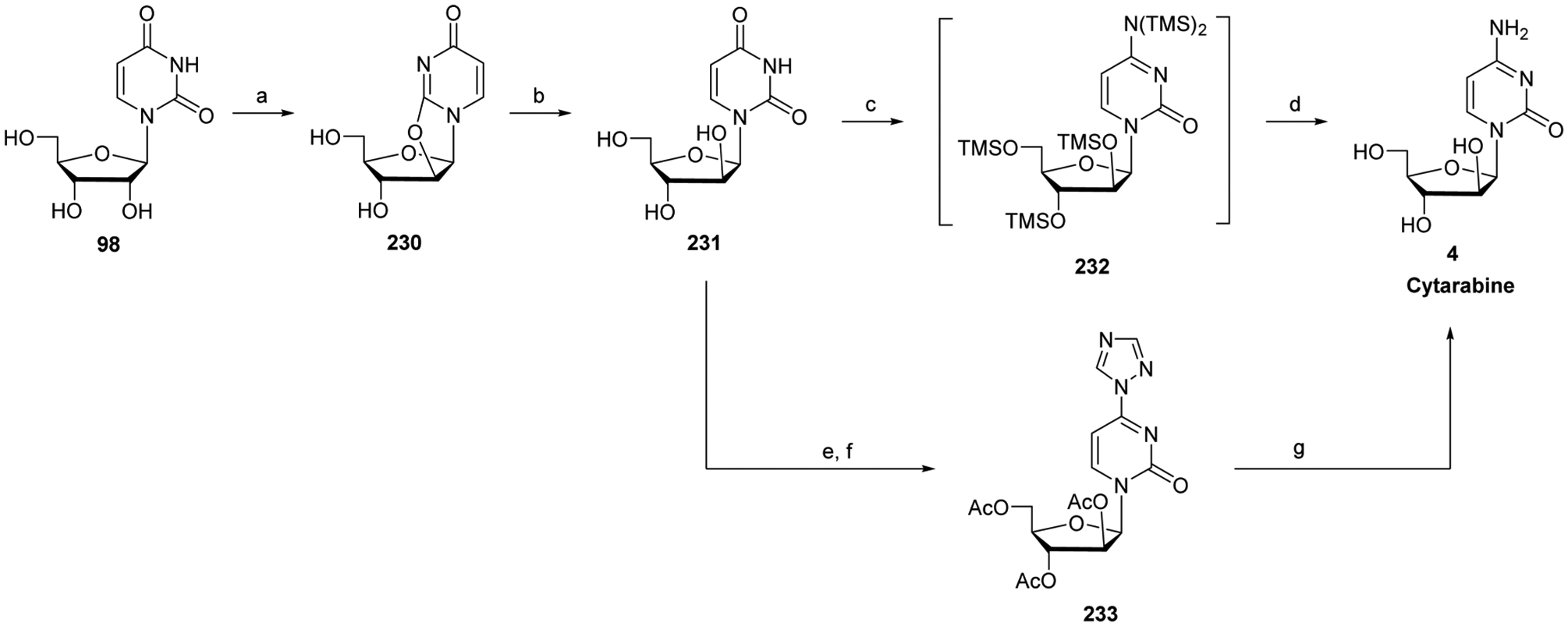


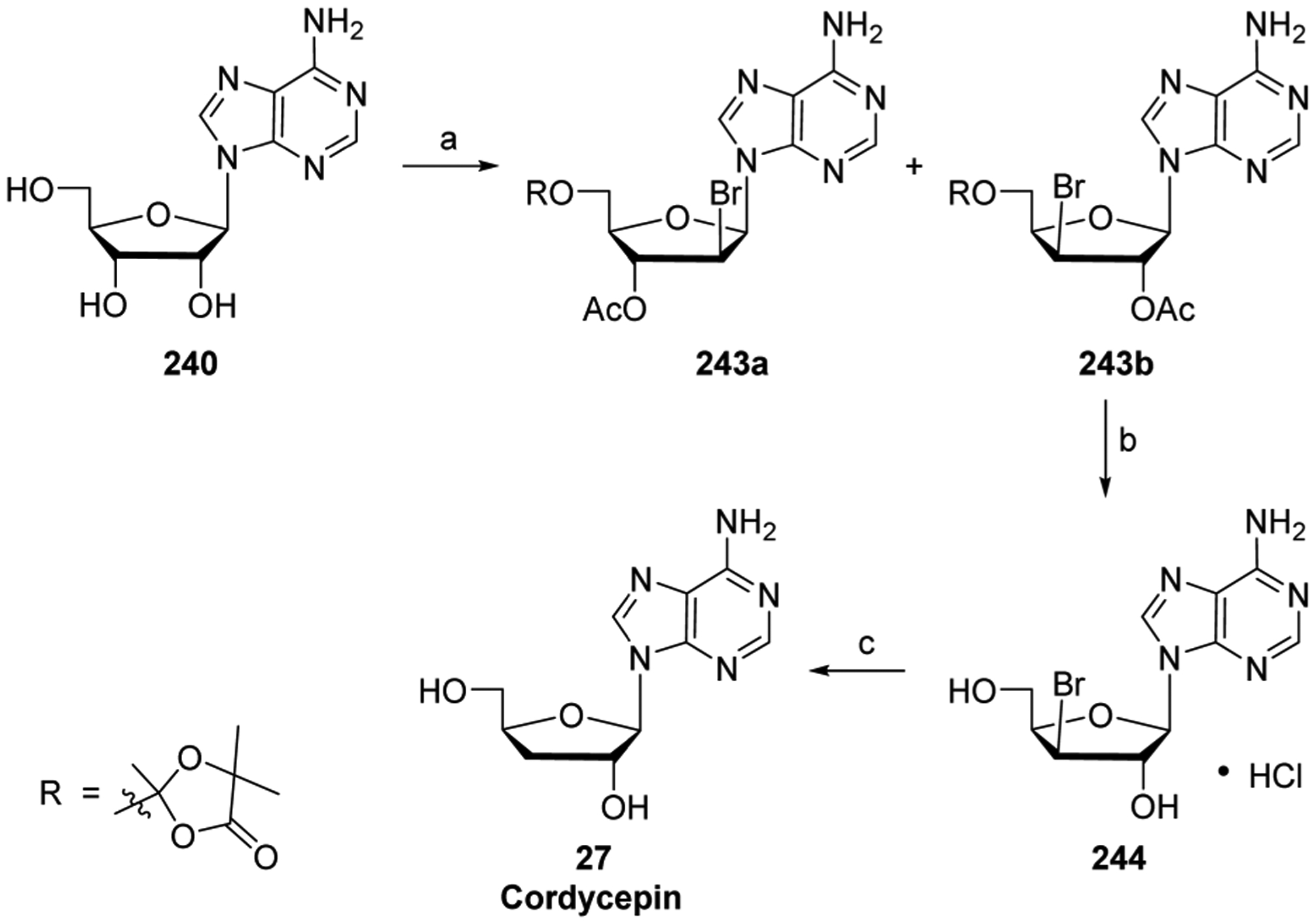




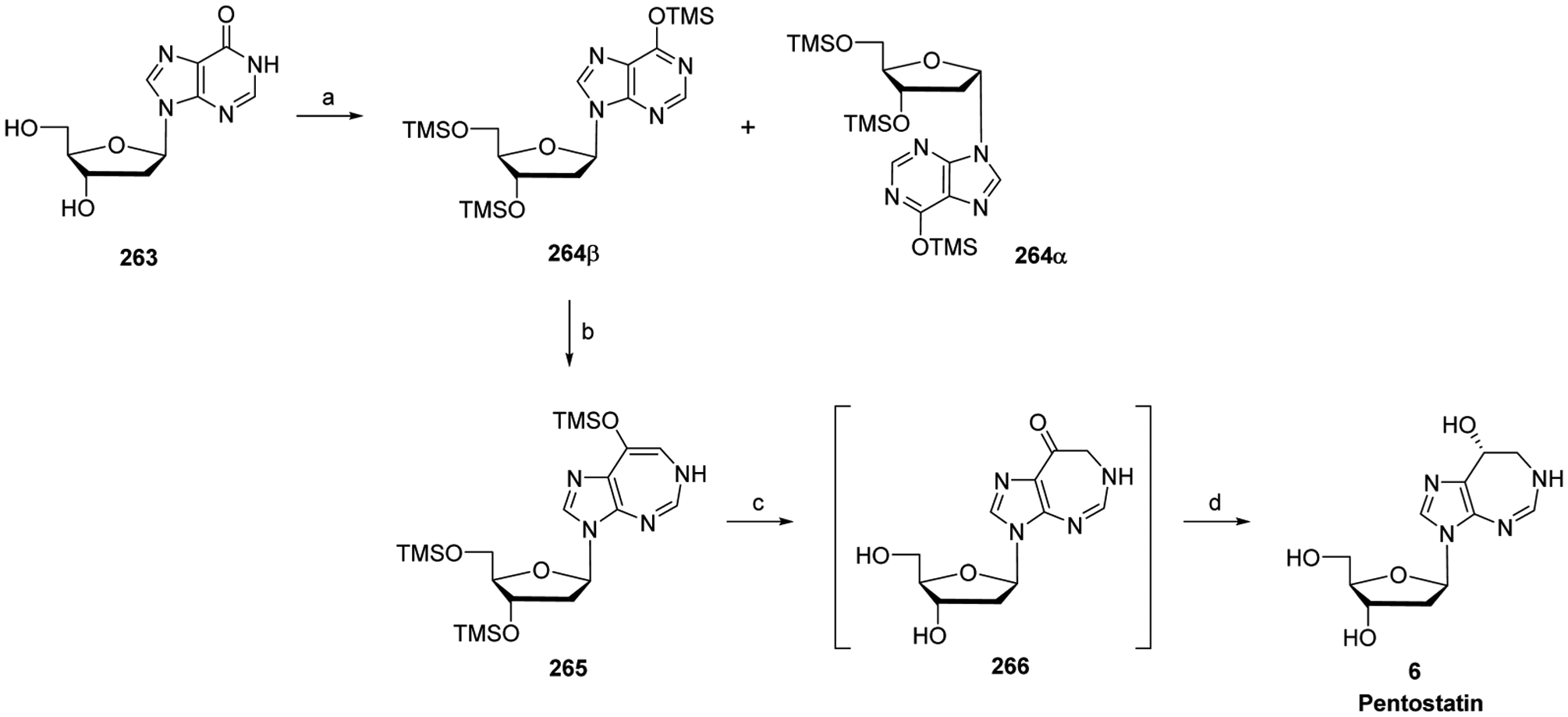






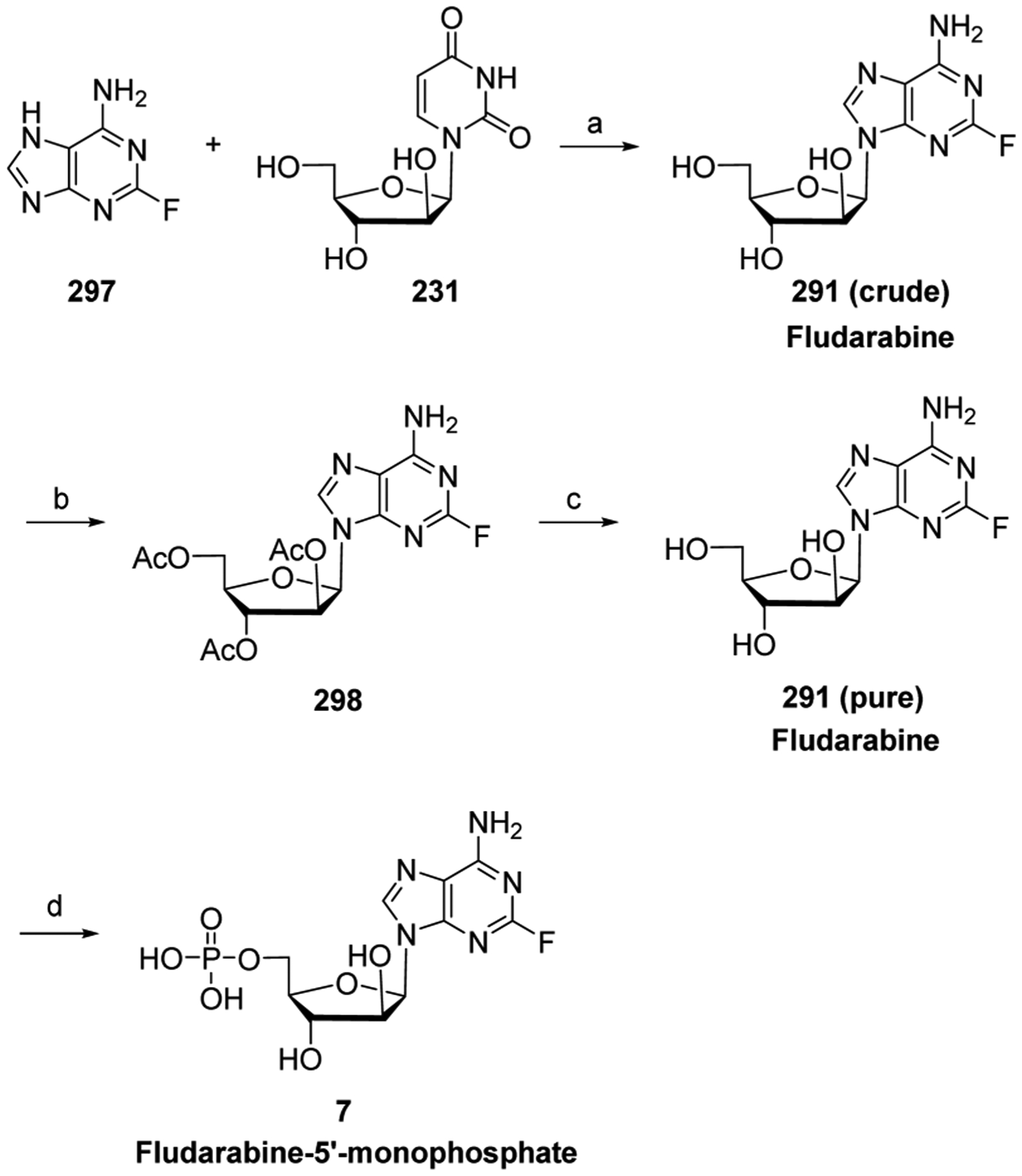
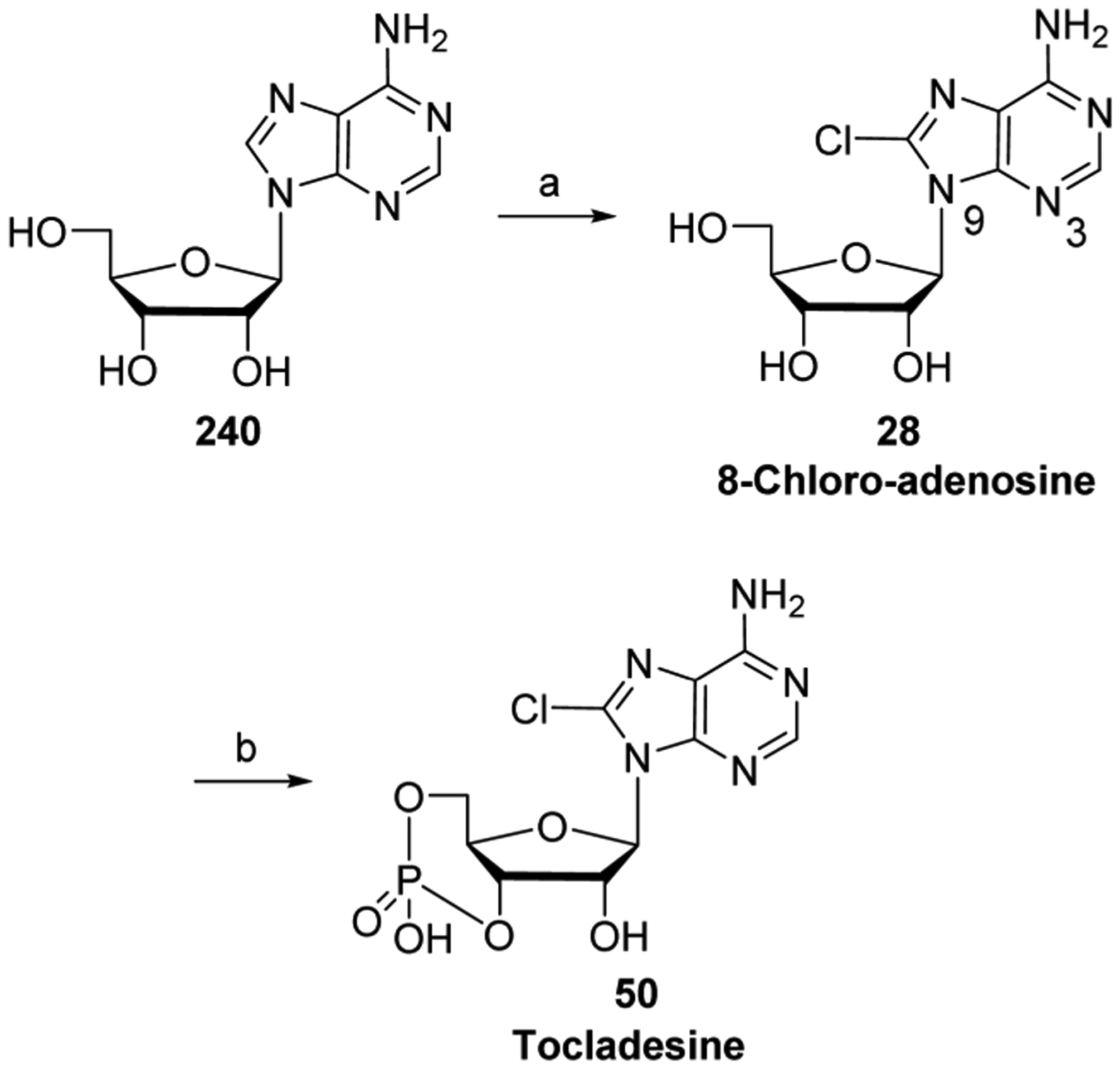
















References
-
- Siegel R; Jemal A Cancer facts & figures 2014; American Cancer Society: 2014; pp 1–68.
-
- Rasool S; Kadla SA; Rasool V; Ganai BA A comparative overview of general risk factors associated with the incidence of colorectal cancer. Tumor Biol 2013, 34 (5), 2469–2476. - PubMed
-
- Zimet GD; Buffler P Prevention of human papillomavirus-related diseases: Impediments to progress. Prev. Med 2013, 57 (5), 407–408. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
