

Downregulation of a GPCR by β-Arrestin2-Mediated Switch from an Endosomal to a TGN Recycling Pathway
- PMID: 27974210
- PMCID: PMC5161243
- DOI: 10.1016/j.celrep.2016.11.050
Downregulation of a GPCR by β-Arrestin2-Mediated Switch from an Endosomal to a TGN Recycling Pathway
Abstract
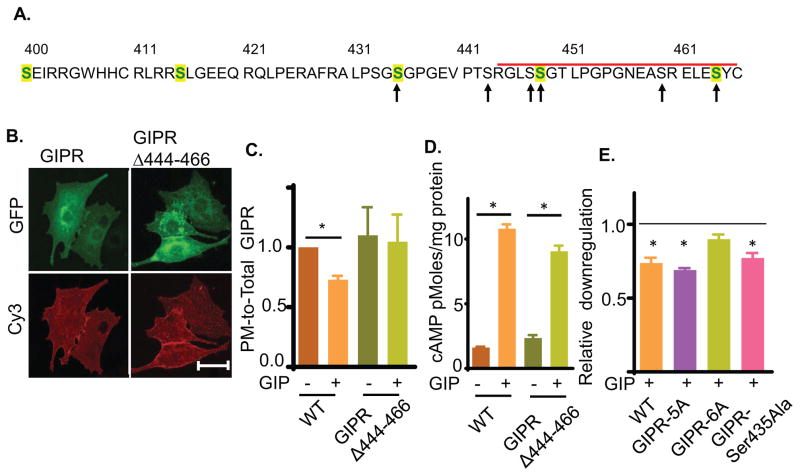
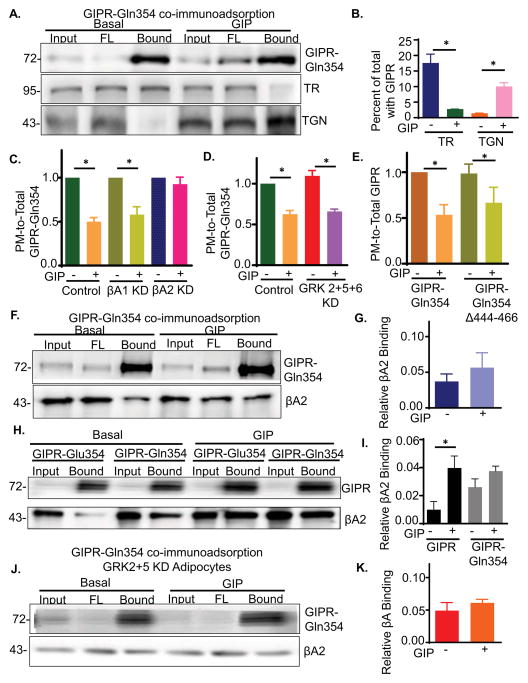
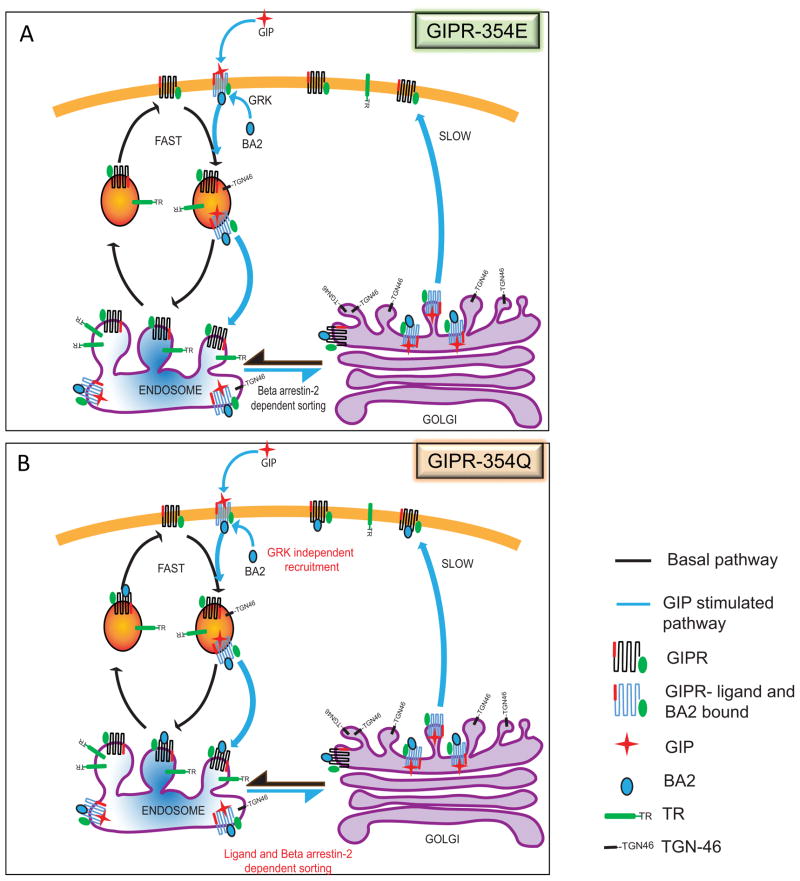
Glucose-dependent insulinotropic polypeptide (GIP) is an incretin hormone involved in nutrient homeostasis. GIP receptor (GIPR) is constitutively internalized and returned to the plasma membrane, atypical behavior for a G-protein-coupled receptor (GPCR). GIP promotes GIPR downregulation from the plasma membrane by inhibiting recycling without affecting internalization. This transient desensitization is achieved by altered intracellular trafficking of activated GIPR. GIP stimulation induces a switch in GIPR recycling from a rapid endosomal to a slow trans-Golgi network (TGN) pathway. GPCR kinases and β-arrestin2 are required for this switch in recycling. A coding sequence variant of GIPR, which has been associated with metabolic alterations, has altered post-activation trafficking characterized by enhanced downregulation and prolonged desensitization. Downregulation of the variant requires β-arrestin2 targeting to the TGN but is independent of GPCR kinases. The single amino acid substitution in the variant biases the receptor to promote GIP-stimulated β-arrestin2 recruitment without receptor phosphorylation, thereby enhancing downregulation.
Keywords: GIP; GIP receptor; GIPR downregulation; GPCR; GPCR trafficking; GRK; beta arrestin; incretin.
Copyright © 2016 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Banting G, Ponnambalam S. TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochimica et biophysica acta. 1997;1355:209–217. - PubMed
-
- Bard F, Malhotra V. The formation of TGN-to-plasma-membrane transport carriers. Annu Rev Cell Dev Biol. 2006;22:439–455. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
