

Murine Electrophysiological Models of Cardiac Arrhythmogenesis
- PMID: 27974512
- PMCID: PMC5539373
- DOI: 10.1152/physrev.00007.2016
Murine Electrophysiological Models of Cardiac Arrhythmogenesis
Abstract
Cardiac arrhythmias can follow disruption of the normal cellular electrophysiological processes underlying excitable activity and their tissue propagation as coherent wavefronts from the primary sinoatrial node pacemaker, through the atria, conducting structures and ventricular myocardium. These physiological events are driven by interacting, voltage-dependent, processes of activation, inactivation, and recovery in the ion channels present in cardiomyocyte membranes. Generation and conduction of these events are further modulated by intracellular Ca2+ homeostasis, and metabolic and structural change. This review describes experimental studies on murine models for known clinical arrhythmic conditions in which these mechanisms were modified by genetic, physiological, or pharmacological manipulation. These exemplars yielded molecular, physiological, and structural phenotypes often directly translatable to their corresponding clinical conditions, which could be investigated at the molecular, cellular, tissue, organ, and whole animal levels. Arrhythmogenesis could be explored during normal pacing activity, regular stimulation, following imposed extra-stimuli, or during progressively incremented steady pacing frequencies. Arrhythmic substrate was identified with temporal and spatial functional heterogeneities predisposing to reentrant excitation phenomena. These could arise from abnormalities in cardiac pacing function, tissue electrical connectivity, and cellular excitation and recovery. Triggering events during or following recovery from action potential excitation could thereby lead to sustained arrhythmia. These surface membrane processes were modified by alterations in cellular Ca2+ homeostasis and energetics, as well as cellular and tissue structural change. Study of murine systems thus offers major insights into both our understanding of normal cardiac activity and its propagation, and their relationship to mechanisms generating clinical arrhythmias.
Copyright © 2017 the American Physiological Society.
Figures





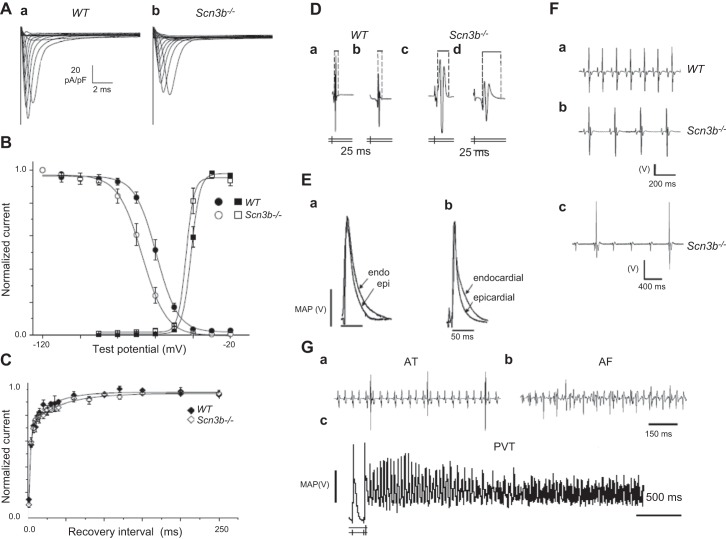
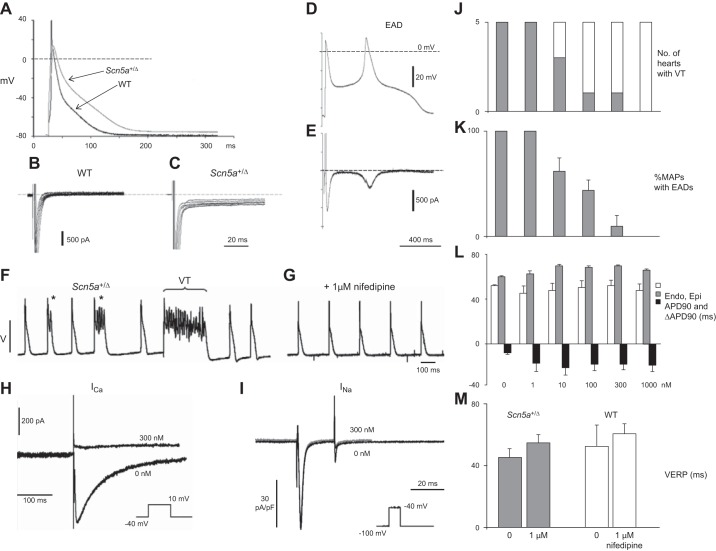

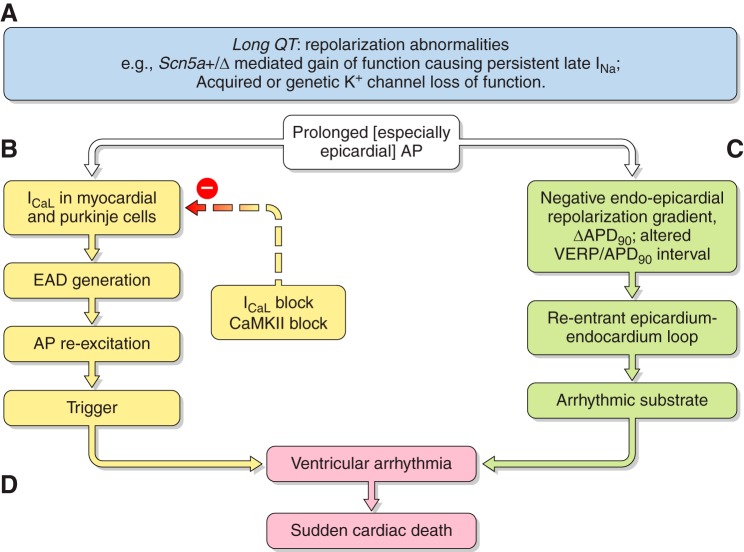
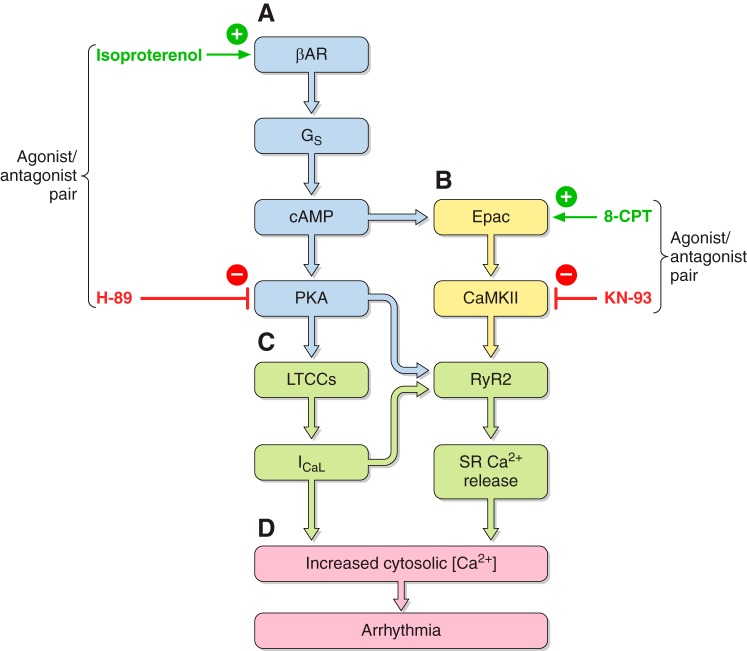
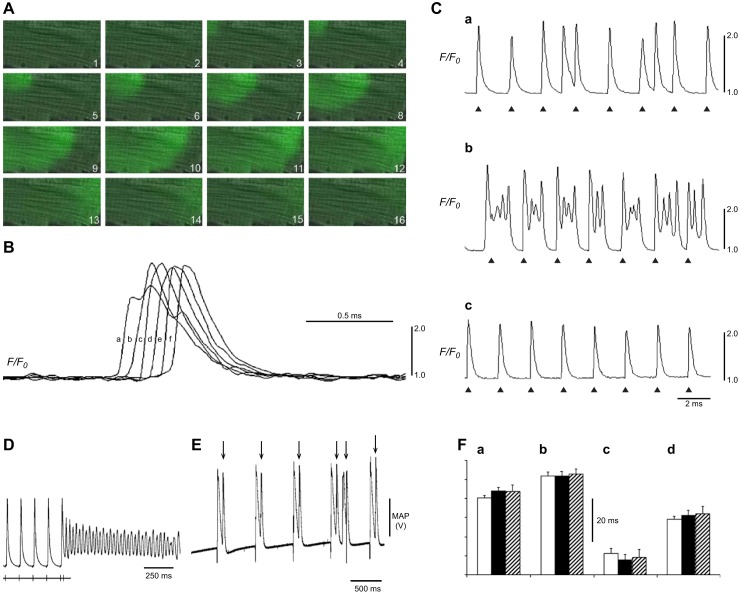
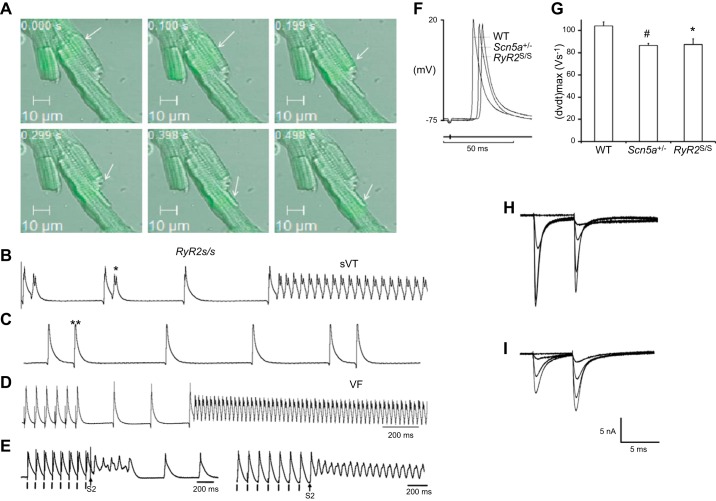
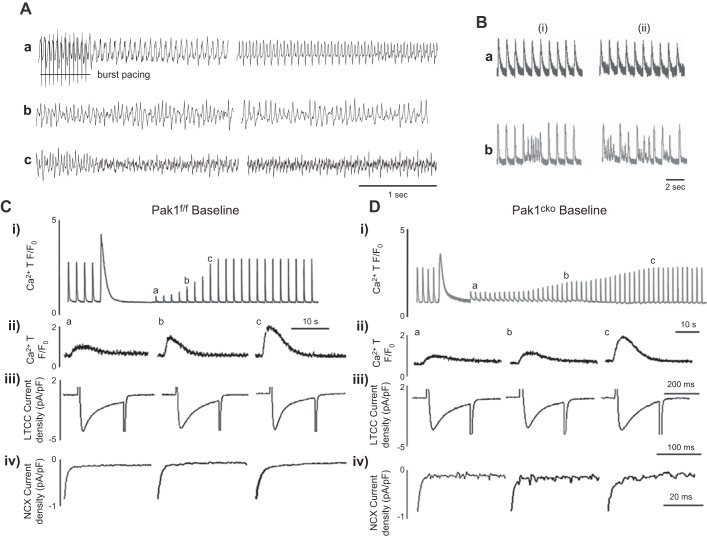
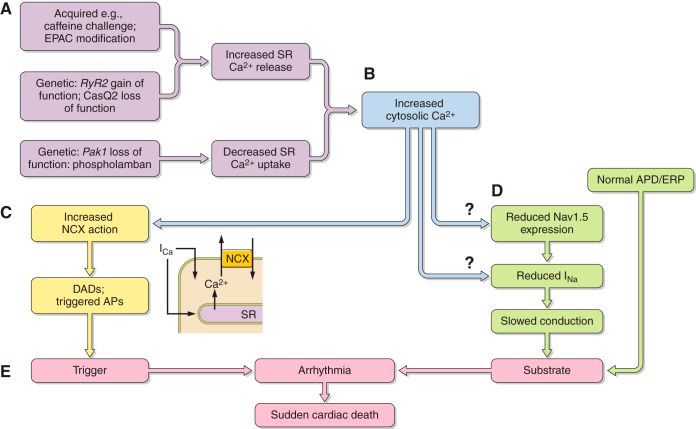
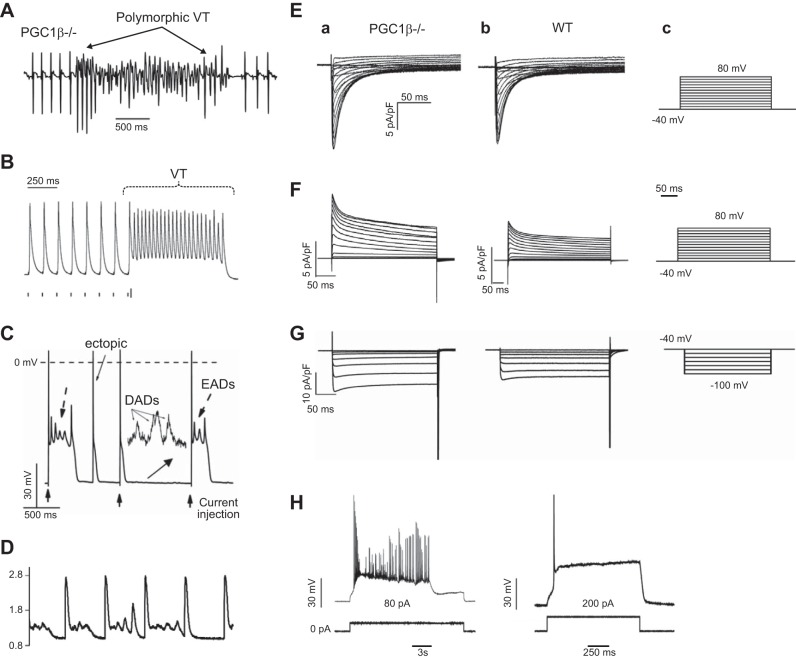
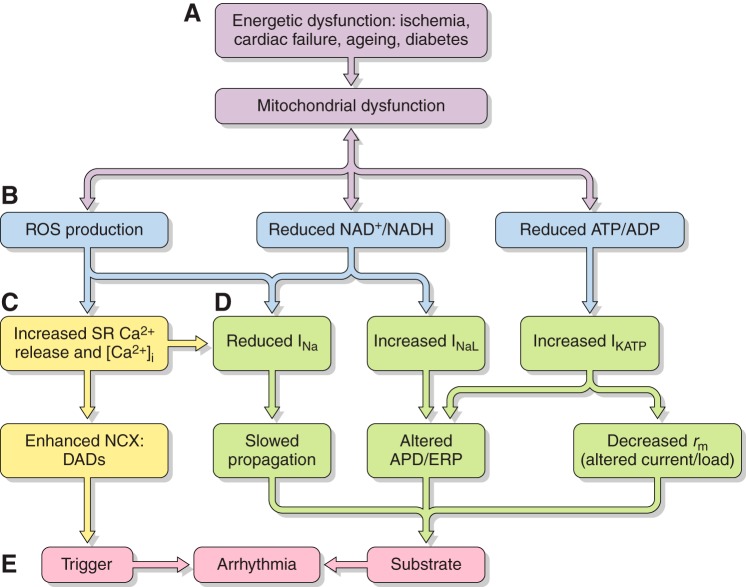
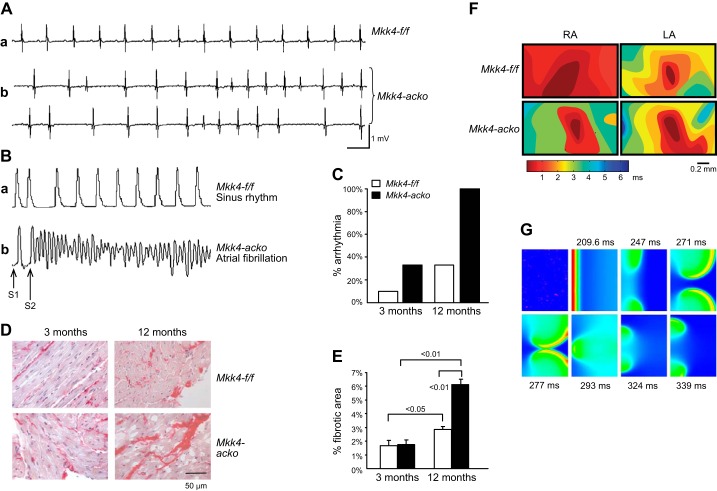
References
-
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–187, 1999. - PubMed
-
- Abriel H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol 48: 2–11, 2010. - PubMed
-
- Adebanjo OA, Anandatheerthavarada HK, Koval AP, Moonga BS, Biswas G, Sun L, Sodam BR, Bevis PJ, Huang CLH, Epstein S, Lai FA, Avadhani NG, Zaidi M. A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol 1: 409–414, 1999. - PubMed
-
- Adebanjo OA, Shankar VS, Pazianas M, Simon BJ, Lai FA, Huang CLH, Zaidi M. Extracellularly applied ruthenium red and cADP ribose elevate cytosolic Ca2+ in isolated rat osteoclasts. Am J Physiol Renal Fluid Electrolyte Physiol 270: F469–F475, 1996. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous