

Myeloperoxidase Enhances Etoposide and Mitoxantrone-Mediated DNA Damage: A Target for Myeloprotection in Cancer Chemotherapy
- PMID: 27974636
- PMCID: PMC5198516
- DOI: 10.1124/mol.116.106054
Myeloperoxidase Enhances Etoposide and Mitoxantrone-Mediated DNA Damage: A Target for Myeloprotection in Cancer Chemotherapy
Abstract
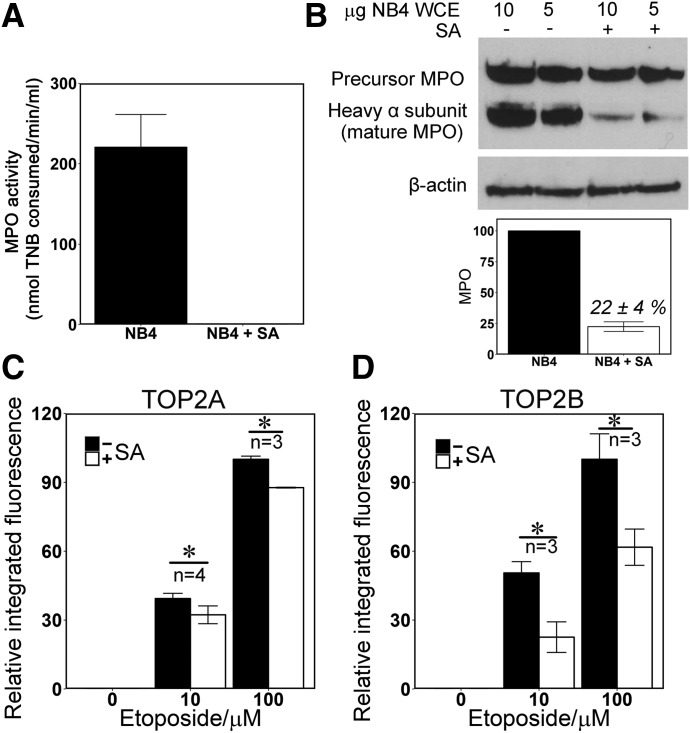
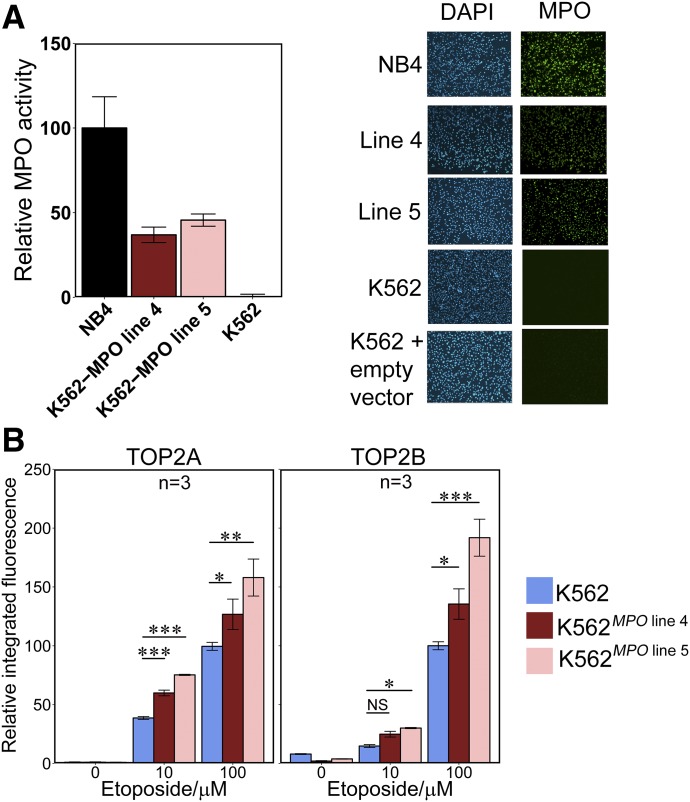
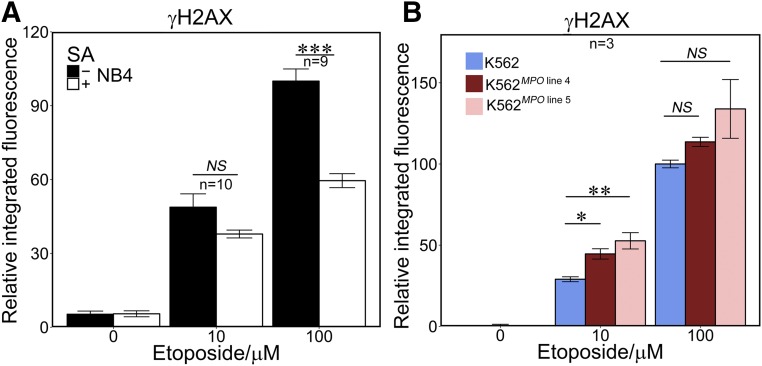
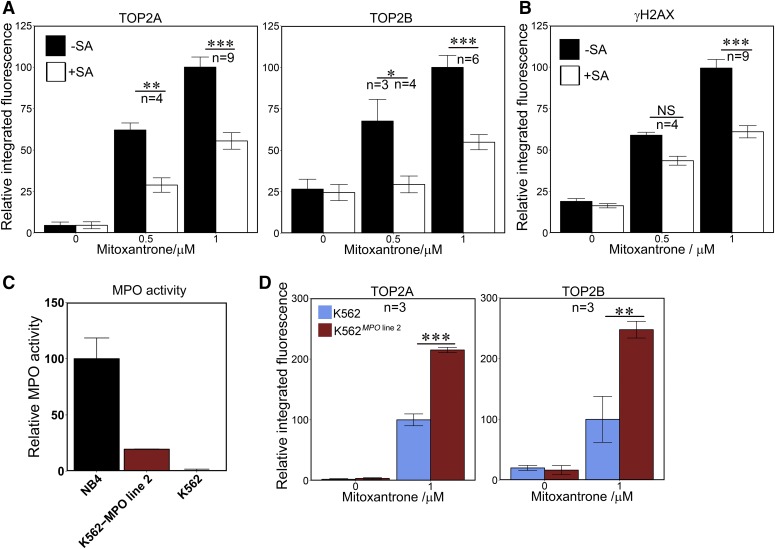
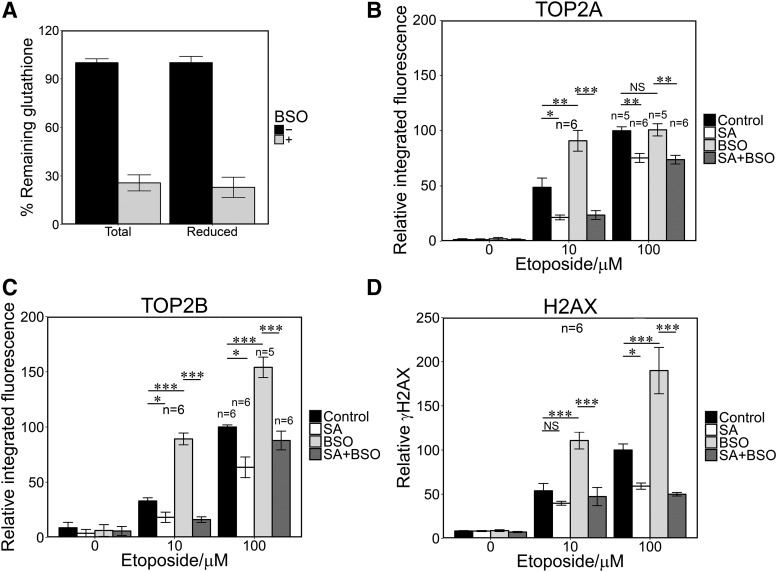
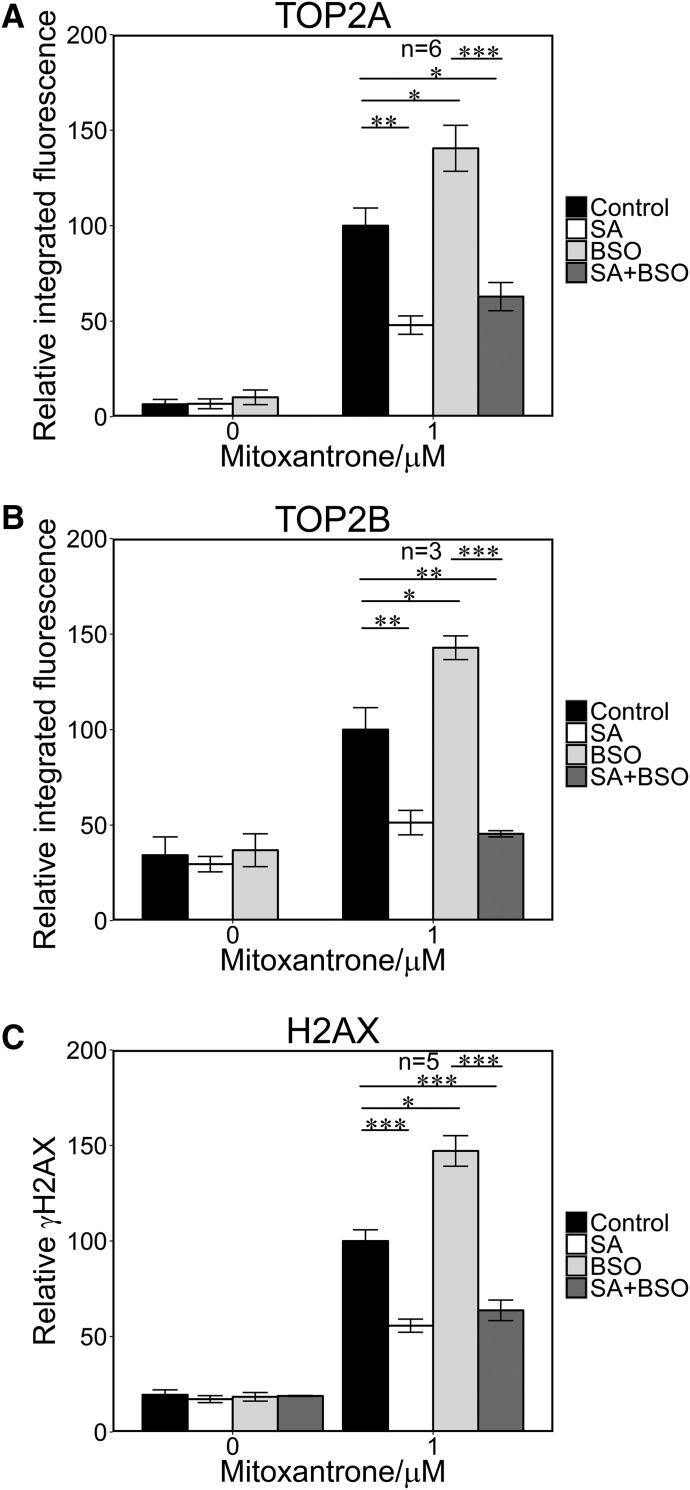
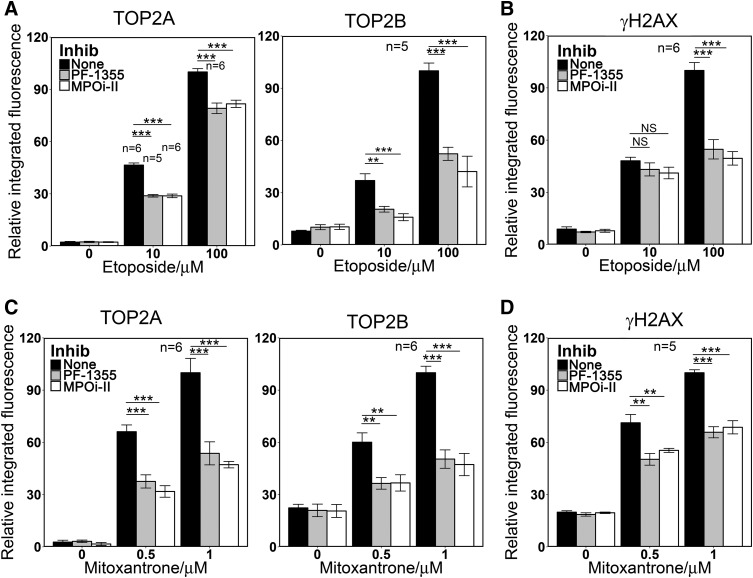
Myeloperoxidase is expressed exclusively in granulocytes and immature myeloid cells and transforms the topoisomerase II (TOP2) poisons etoposide and mitoxantrone to chemical forms that have altered DNA damaging properties. TOP2 poisons are valuable and widely used anticancer drugs, but they are associated with the occurrence of secondary acute myeloid leukemias. These factors have led to the hypothesis that myeloperoxidase inhibition could protect hematopoietic cells from TOP2 poison-mediated genotoxic damage and, therefore, reduce the rate of therapy-related leukemia. We show here that myeloperoxidase activity leads to elevated accumulation of etoposide- and mitoxantrone-induced TOP2A and TOP2B-DNA covalent complexes in cells, which are converted to DNA double-strand breaks. For both drugs, the effect of myeloperoxidase activity was greater for TOP2B than for TOP2A. This is a significant finding because TOP2B has been linked to genetic damage associated with leukemic transformation, including etoposide-induced chromosomal breaks at the MLL and RUNX1 loci. Glutathione depletion, mimicking in vivo conditions experienced during chemotherapy treatment, elicited further MPO-dependent increase in TOP2A and especially TOP2B-DNA complexes and DNA double-strand break formation. Together these results support targeting myeloperoxidase activity to reduce genetic damage leading to therapy-related leukemia, a possibility that is enhanced by the recent development of novel specific myeloperoxidase inhibitors for use in inflammatory diseases involving neutrophil infiltration.
Copyright © 2016 The Author(s).
Figures
References
-
- Allan JM, Travis LB. (2005) Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer 5:943–955. - PubMed
-
- Austin CA, Marsh KL. (1998) Eukaryotic DNA topoisomerase II β. BioEssays 20:215–226. - PubMed
-
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
