

The recurrent architecture of tumour initiation, progression and drug sensitivity
- PMID: 27977008
- PMCID: PMC5541669
- DOI: 10.1038/nrc.2016.124
The recurrent architecture of tumour initiation, progression and drug sensitivity
Abstract
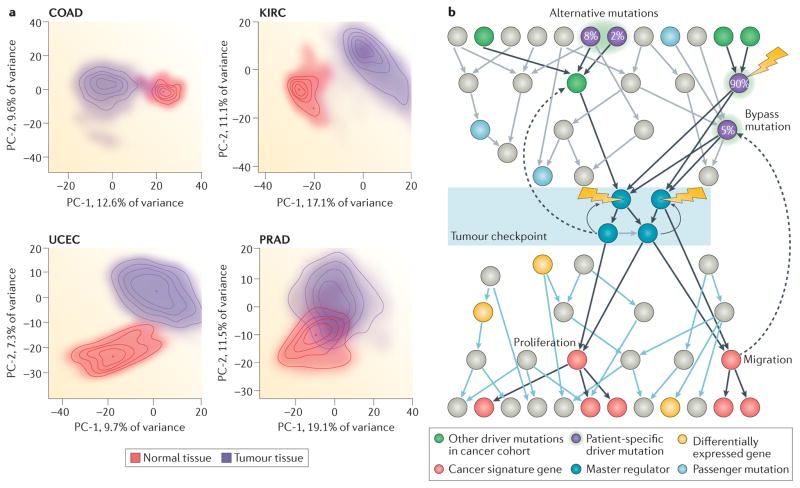
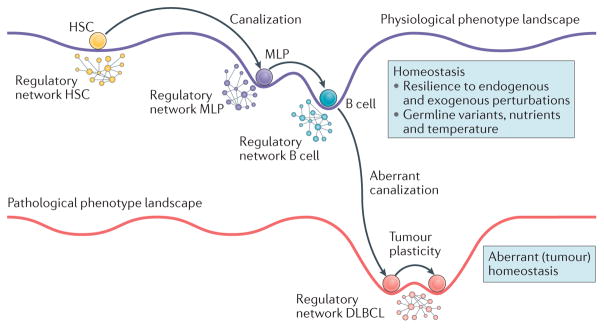
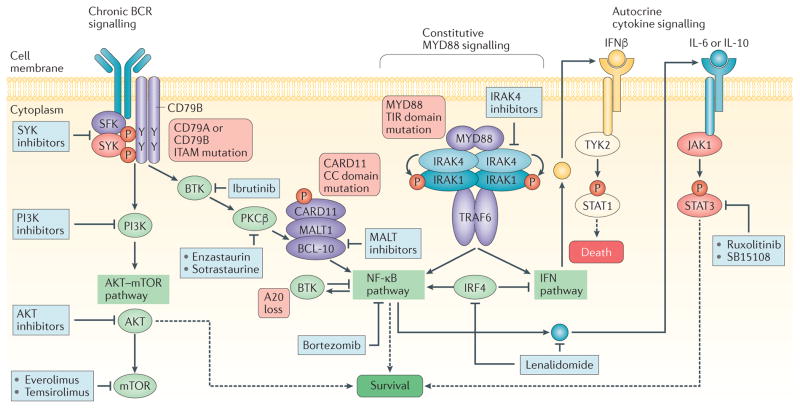
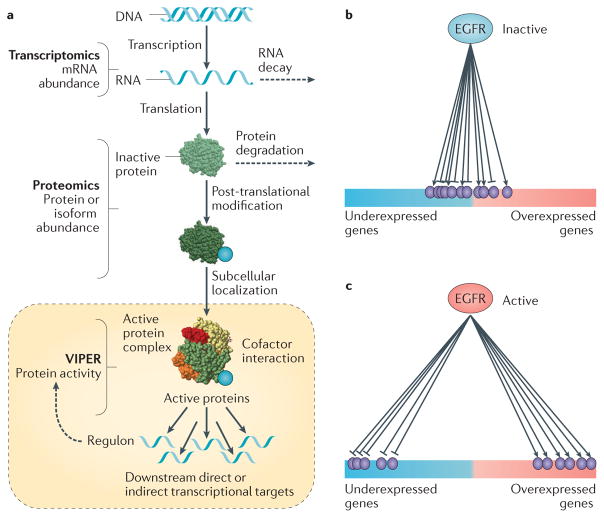
Recent studies across multiple tumour types are starting to reveal a recurrent regulatory architecture in which genomic alterations cluster upstream of functional master regulator (MR) proteins, the aberrant activity of which is both necessary and sufficient to maintain tumour cell state. These proteins form small, hyperconnected and autoregulated modules (termed tumour checkpoints) that are increasingly emerging as optimal biomarkers and therapeutic targets. Crucially, as their activity is mostly dysregulated in a post-translational manner, rather than by mutations in their corresponding genes or by differential expression, the identification of MR proteins by conventional methods is challenging. In this Opinion article, we discuss novel methods for the systematic analysis of MR proteins and of the modular regulatory architecture they implement, including their use as a valuable reductionist framework to study the genetic heterogeneity of human disease and to drive key translational applications.
Conflict of interest statement
The authors declare
A. C is founder of DarwinHealth, Inc. M. J. A. has been employed by DarwinHealth, Inc. since March 2016.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
