

Loss of RMI2 Increases Genome Instability and Causes a Bloom-Like Syndrome
- PMID: 27977684
- PMCID: PMC5157948
- DOI: 10.1371/journal.pgen.1006483
Loss of RMI2 Increases Genome Instability and Causes a Bloom-Like Syndrome
Abstract
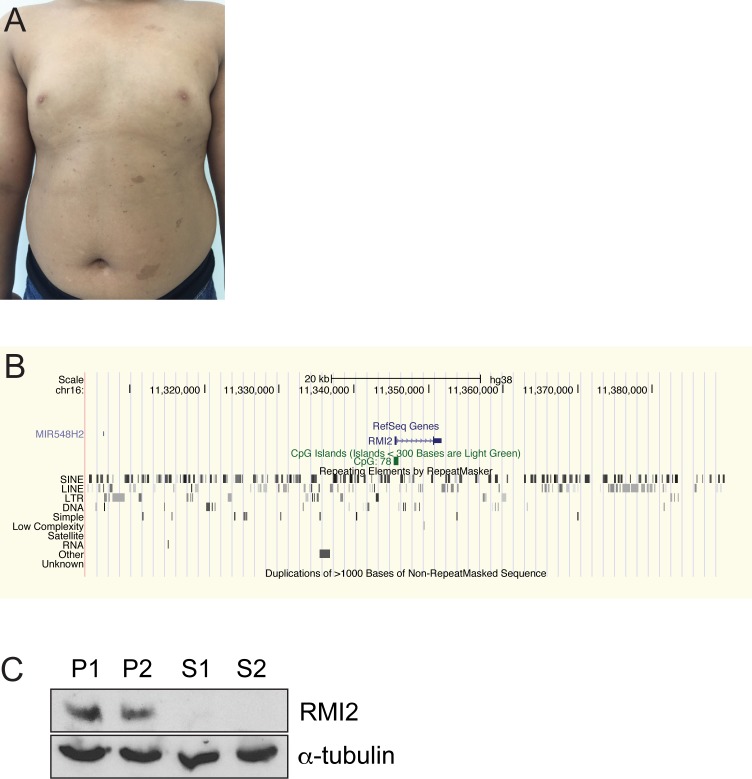
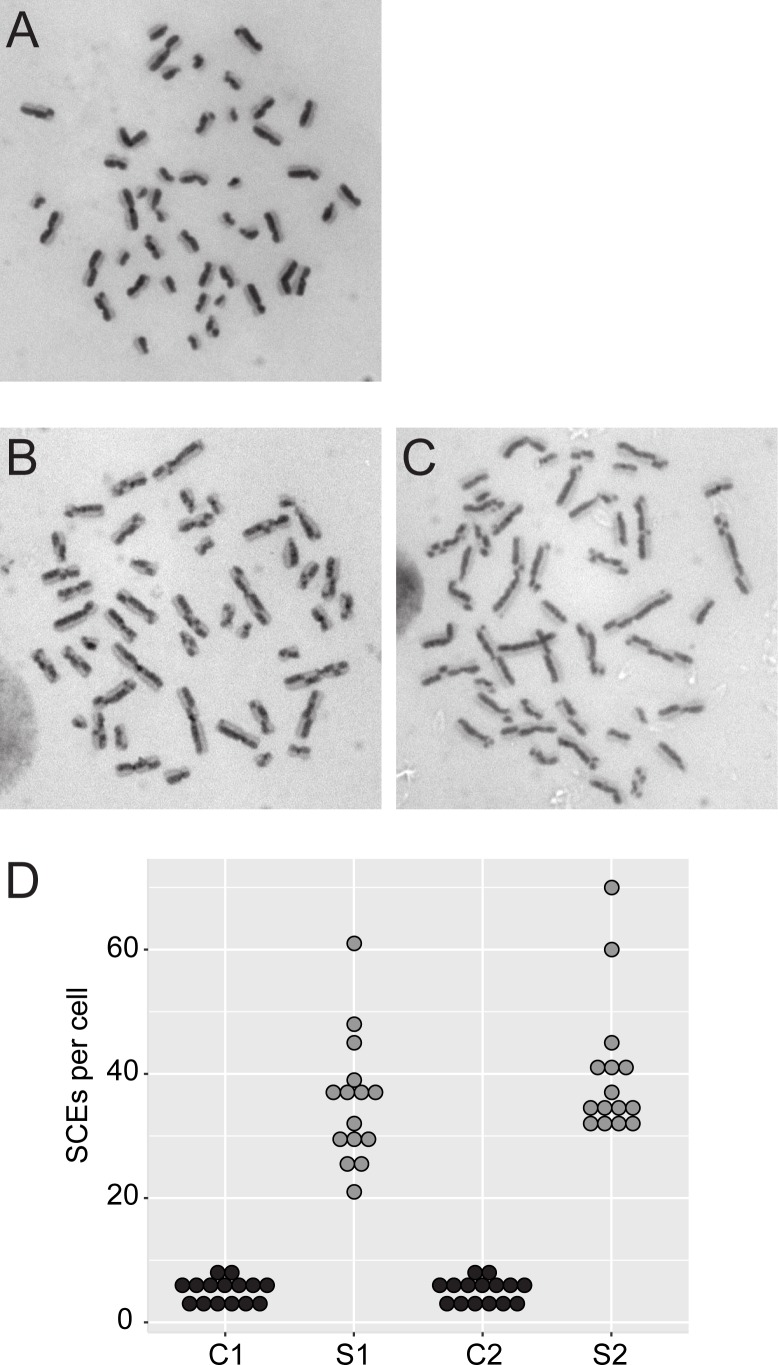
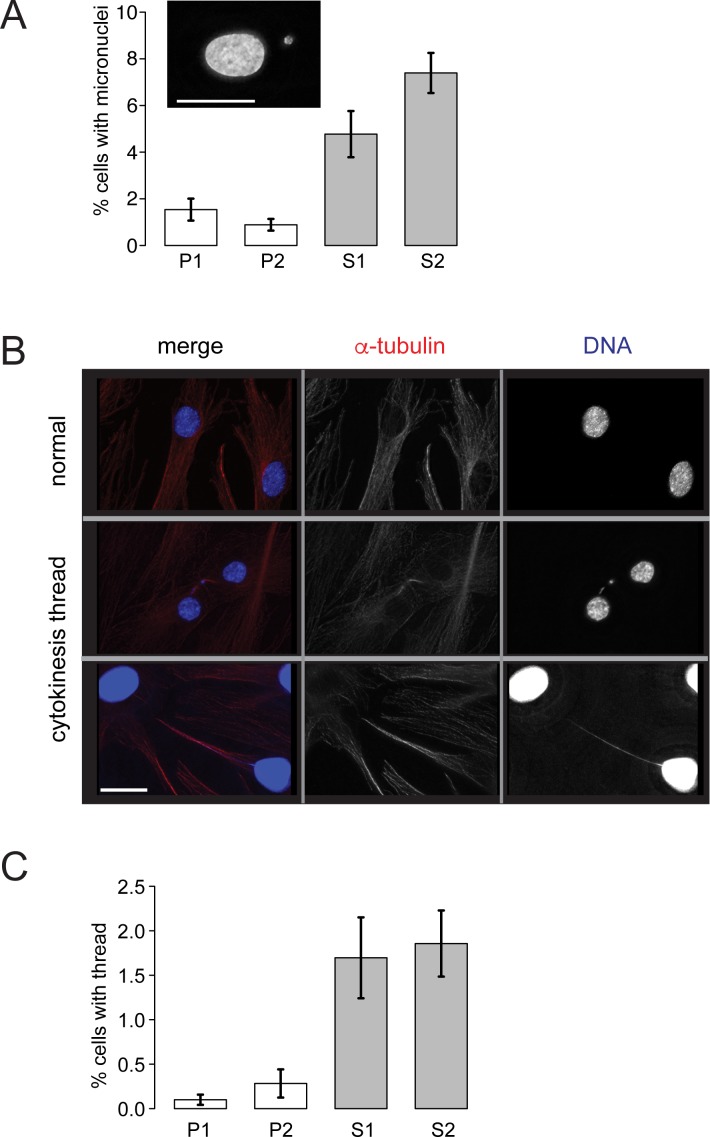
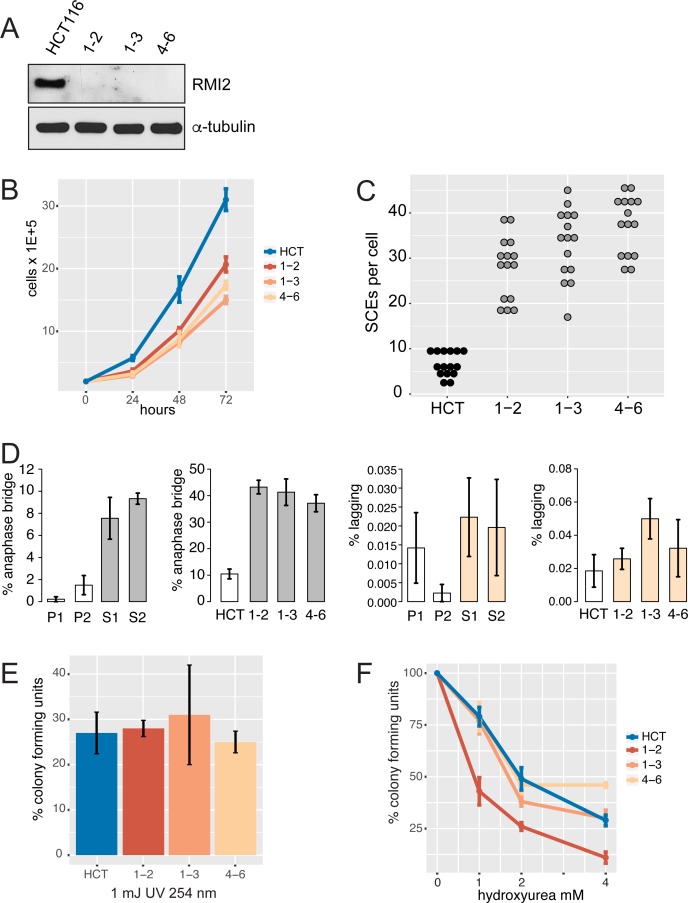
Bloom syndrome is a recessive human genetic disorder with features of genome instability, growth deficiency and predisposition to cancer. The only known causative gene is the BLM helicase that is a member of a protein complex along with topoisomerase III alpha, RMI1 and 2, which maintains replication fork stability and dissolves double Holliday junctions to prevent genome instability. Here we report the identification of a second gene, RMI2, that is deleted in affected siblings with Bloom-like features. Cells from homozygous individuals exhibit elevated rates of sister chromatid exchange, anaphase DNA bridges and micronuclei. Similar genome and chromosome instability phenotypes are observed in independently derived RMI2 knockout cells. In both patient and knockout cell lines reduced localisation of BLM to ultra fine DNA bridges and FANCD2 at foci linking bridges are observed. Overall, loss of RMI2 produces a partially active BLM complex with mild features of Bloom syndrome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
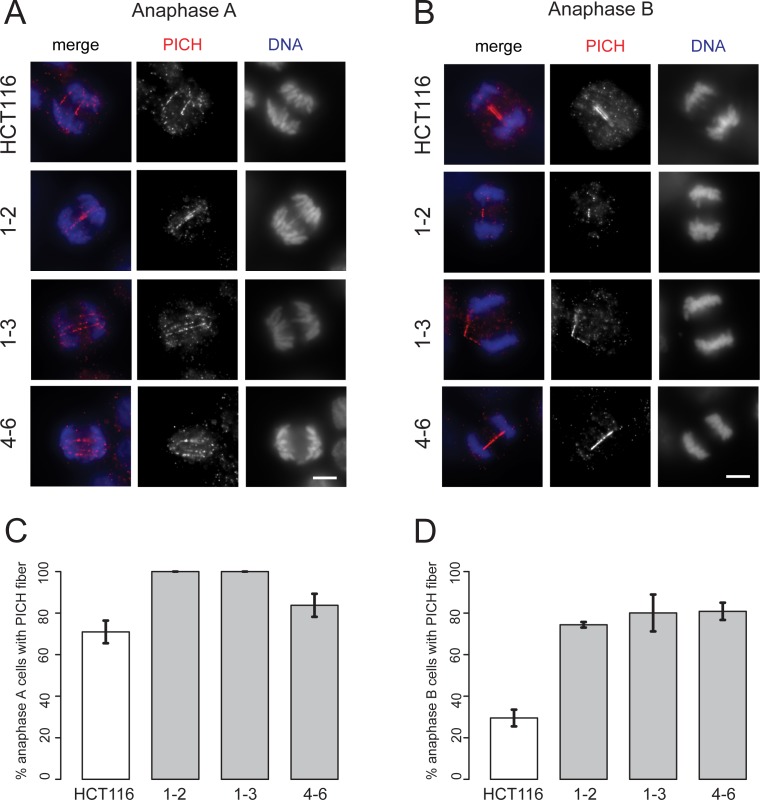
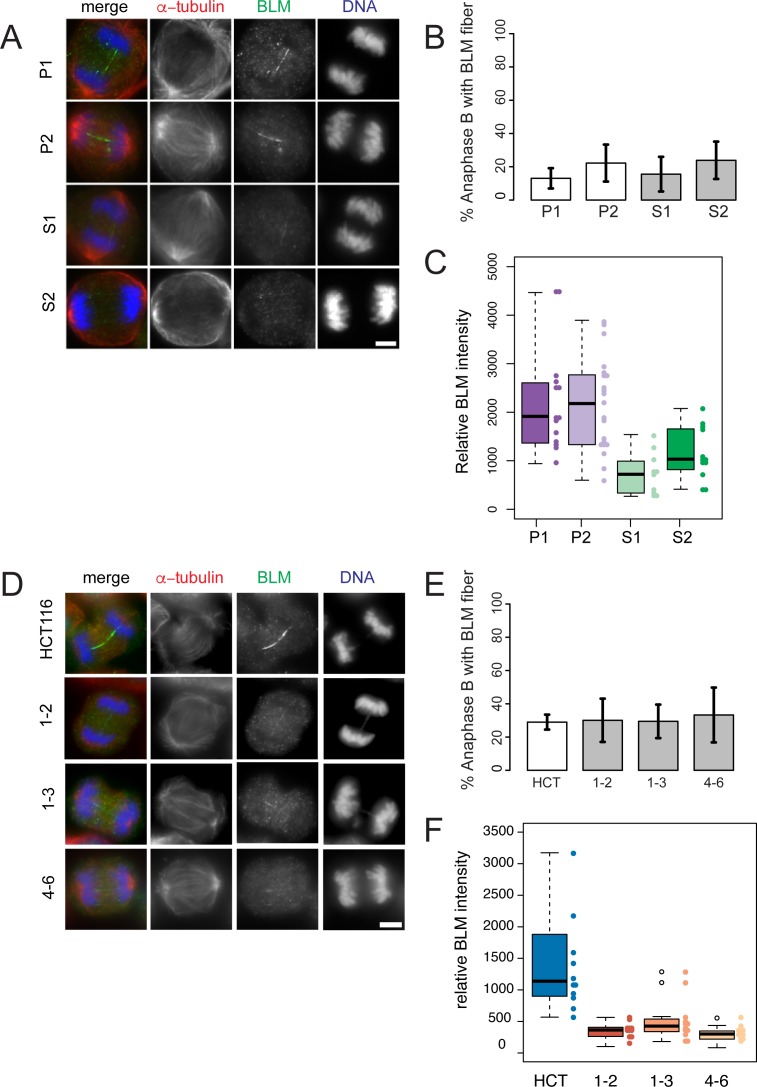
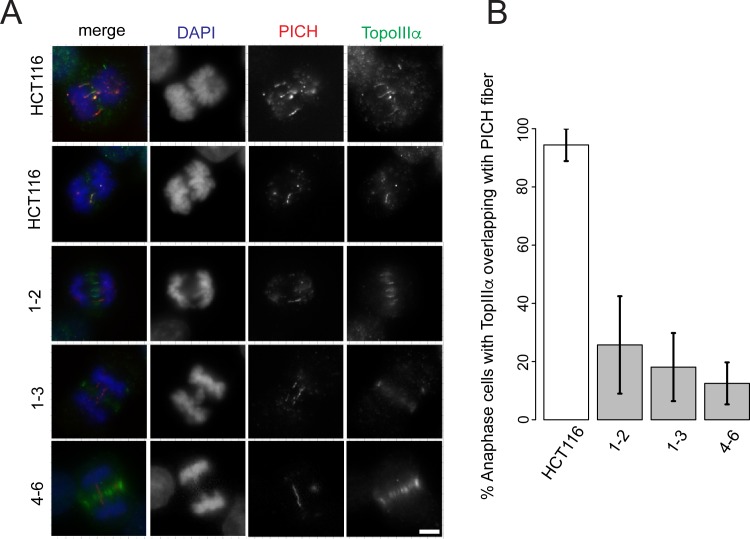
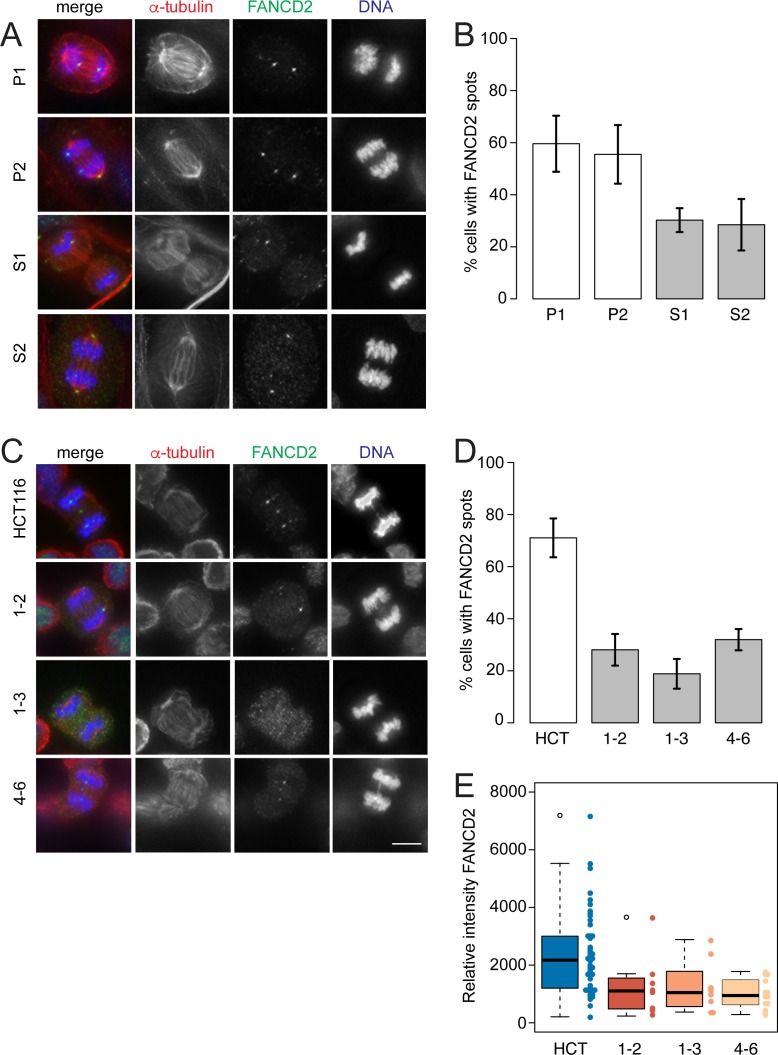
References
-
- German J. Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine (Baltimore). 1993;72: 393–406. - PubMed
-
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83: 655–666. - PubMed
-
- Johnson FB, Lombard DB, Neff NF, Mastrangelo MA, Dewolf W, Ellis NA, et al. Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res. 2000;60: 1162–1167. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
