

GSK-3β/NFAT Signaling Is Involved in Testosterone-Induced Cardiac Myocyte Hypertrophy
- PMID: 27977752
- PMCID: PMC5158037
- DOI: 10.1371/journal.pone.0168255
GSK-3β/NFAT Signaling Is Involved in Testosterone-Induced Cardiac Myocyte Hypertrophy
Abstract
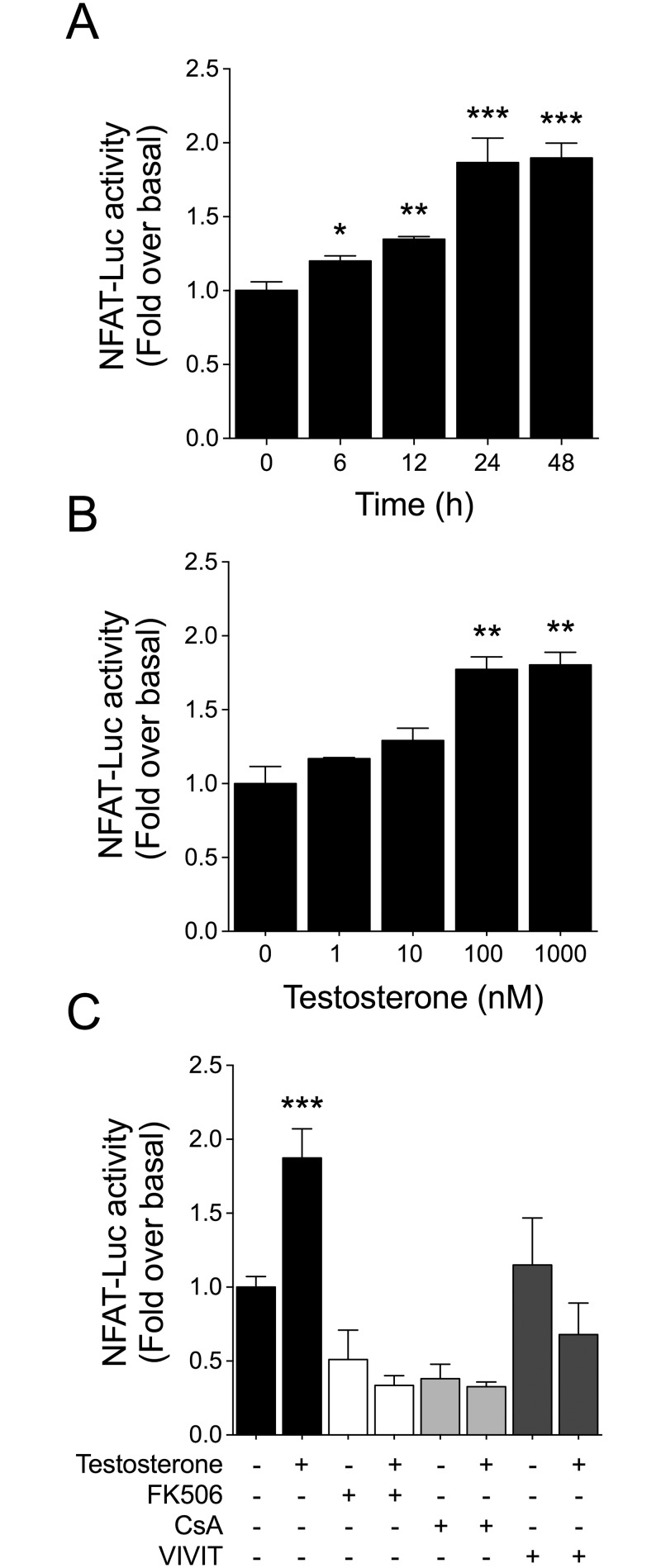
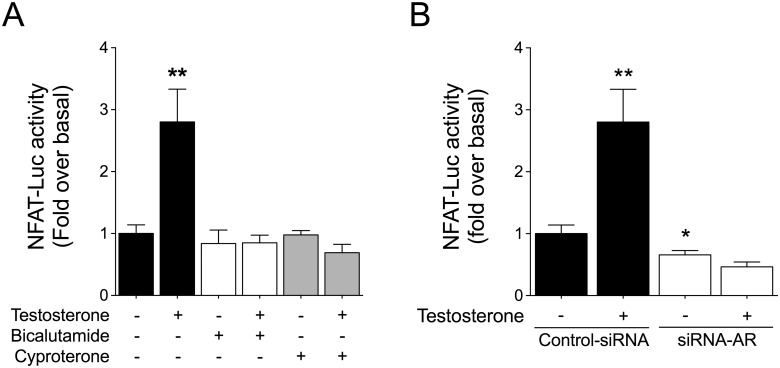
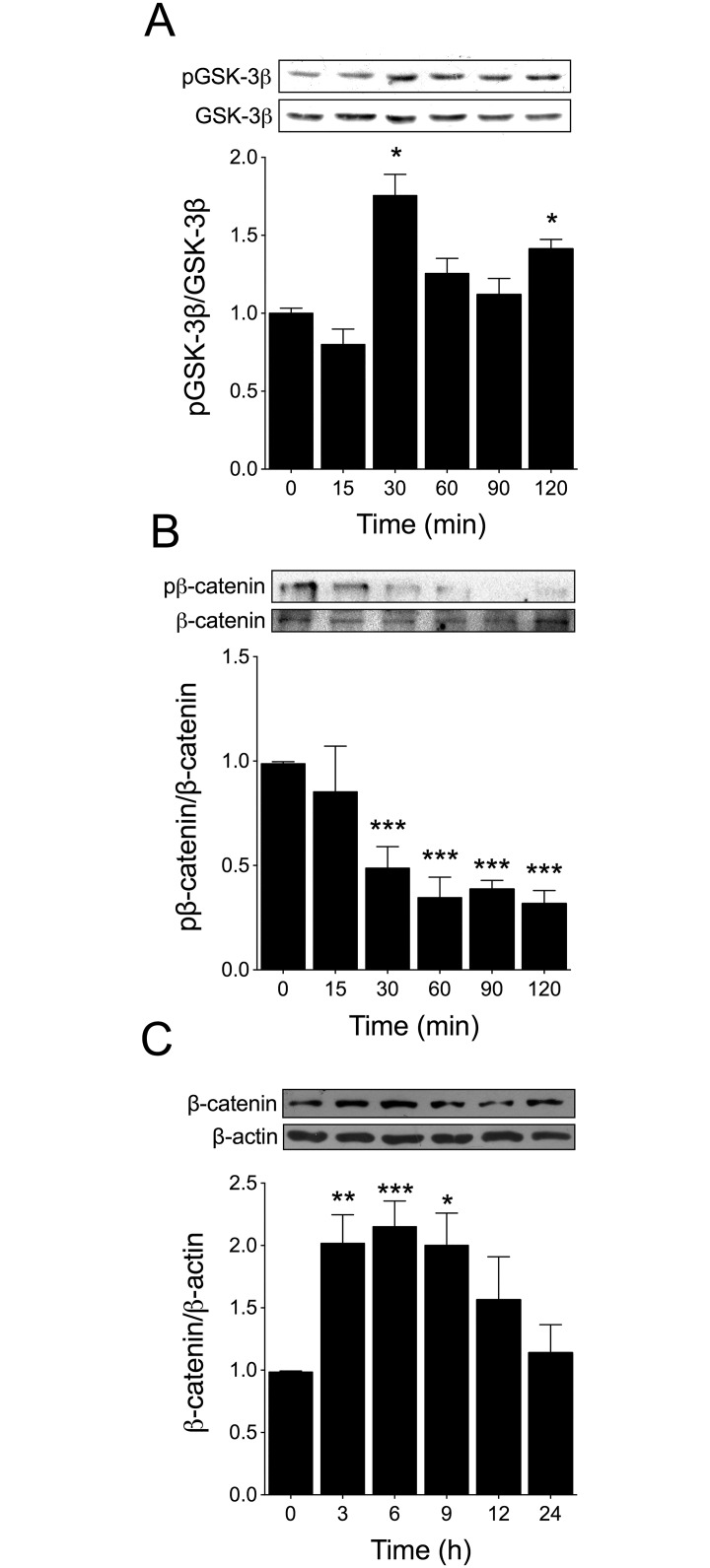
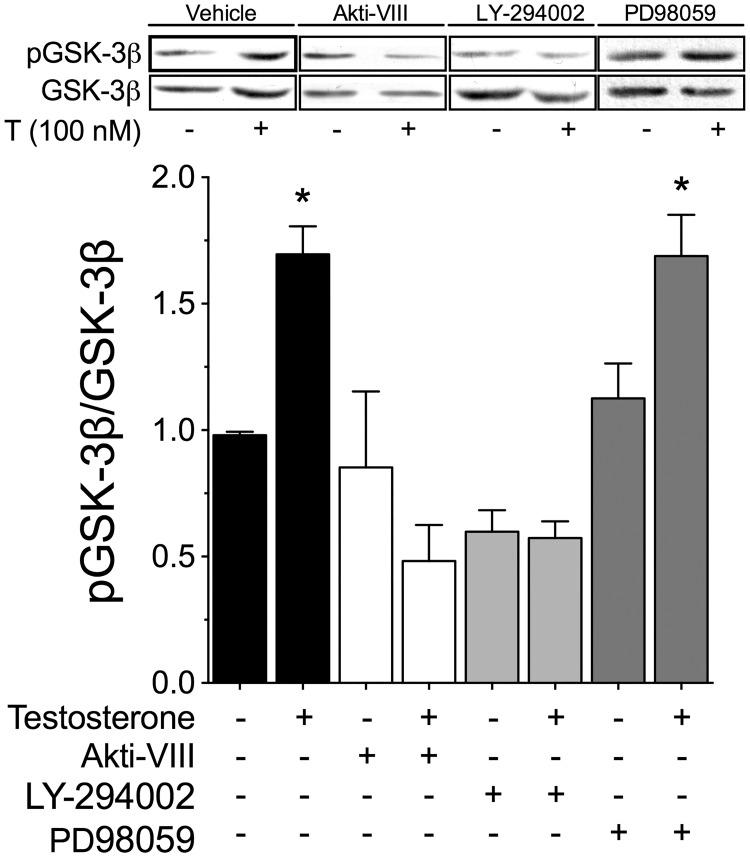
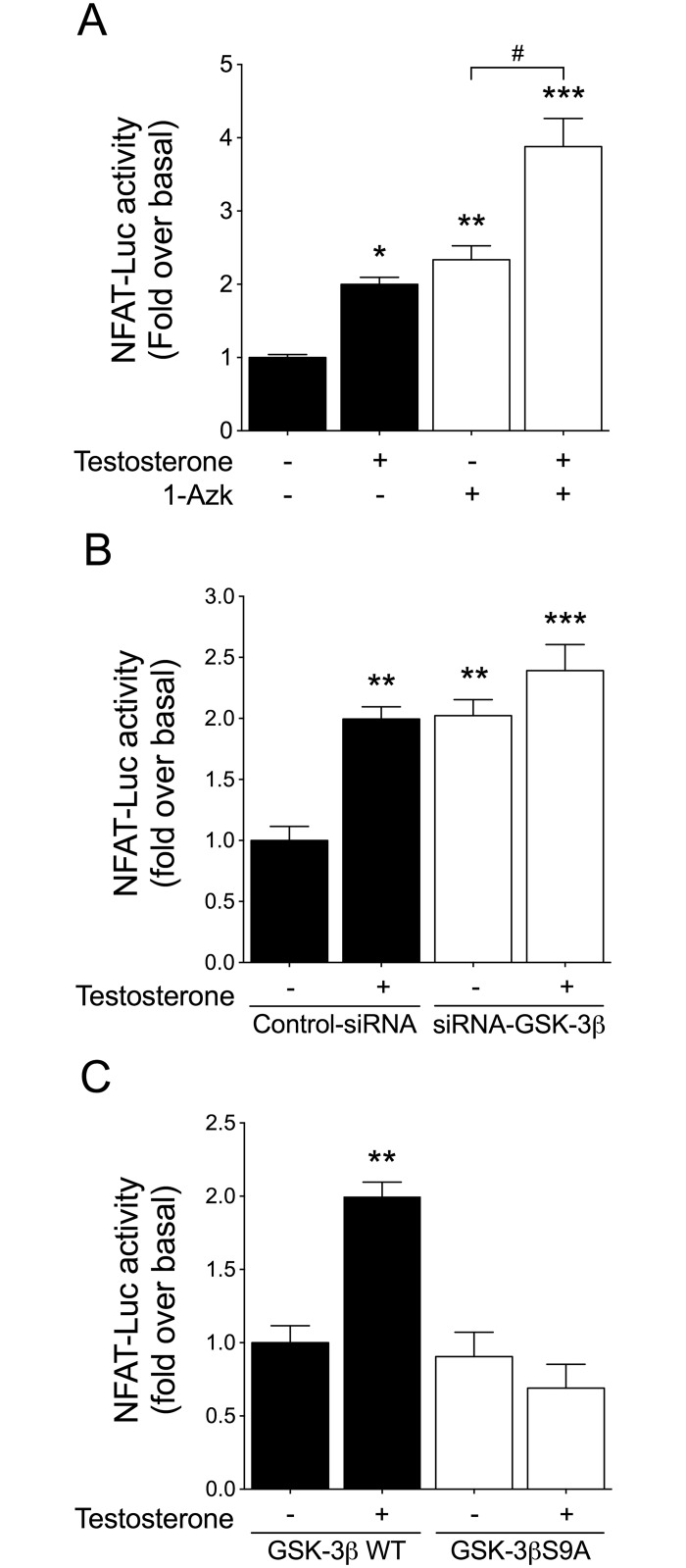
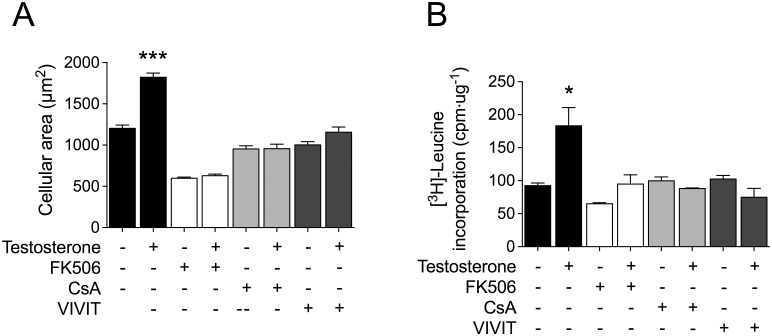
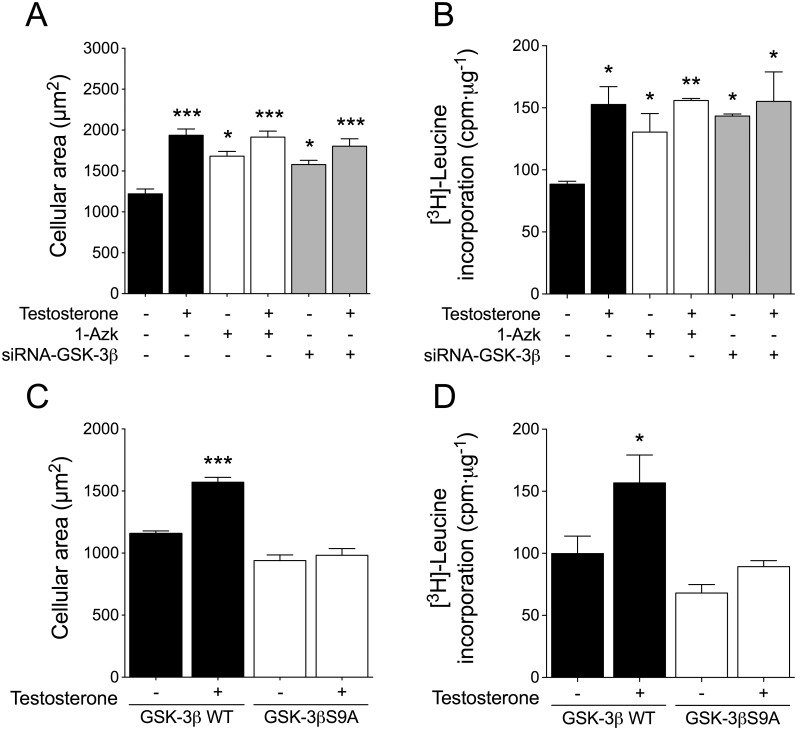
Testosterone induces cardiac hypertrophy through a mechanism that involves a concerted crosstalk between cytosolic and nuclear signaling pathways. Nuclear factor of activated T-cells (NFAT) is associated with the promotion of cardiac hypertrophy, glycogen synthase kinase-3β (GSK-3β) is considered to function as a negative regulator, mainly by modulating NFAT activity. However, the role played by calcineurin-NFAT and GSK-3β signaling in testosterone-induced cardiac hypertrophy has remained unknown. Here, we determined that testosterone stimulates cardiac myocyte hypertrophy through NFAT activation and GSK-3β inhibition. Testosterone increased the activity of NFAT-luciferase (NFAT-Luc) in a time- and dose-dependent manner, with the activity peaking after 24 h of stimulation with 100 nM testosterone. NFAT-Luc activity induced by testosterone was blocked by the calcineurin inhibitors FK506 and cyclosporine A and by 11R-VIVIT, a specific peptide inhibitor of NFAT. Conversely, testosterone inhibited GSK-3β activity as determined by increased GSK-3β phosphorylation at Ser9 and β-catenin protein accumulation, and also by reduction in β-catenin phosphorylation at residues Ser33, Ser37, and Thr41. GSK-3β inhibition with 1-azakenpaullone or a GSK-3β-targeting siRNA increased NFAT-Luc activity, whereas overexpression of a constitutively active GSK-3β mutant (GSK-3βS9A) inhibited NFAT-Luc activation mediated by testosterone. Testosterone-induced cardiac myocyte hypertrophy was established by increased cardiac myocyte size and [3H]-leucine incorporation (as a measurement of cellular protein synthesis). Calcineurin-NFAT inhibition abolished and GSK-3β inhibition promoted the hypertrophy stimulated by testosterone. GSK-3β activation by GSK-3βS9A blocked the increase of hypertrophic markers induced by testosterone. Moreover, inhibition of intracellular androgen receptor prevented testosterone-induced NFAT-Luc activation. Collectively, these results suggest that cardiac myocyte hypertrophy induced by testosterone involves a cooperative mechanism that links androgen signaling with the recruitment of NFAT through calcineurin activation and GSK-3β inhibition.
Conflict of interest statement
The authors have declared that no competing interests exist. I confirm that my commercial affiliation does not alter our adherence to all PLOS policies on sharing data and materials.
Figures
References
-
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109(13): 1580–1589. 10.1161/01.CIR.0000120390.68287.BB - DOI - PubMed
-
- Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. Faseb J. 1991;5(15): 3037–3046. - PubMed
-
- Campbell SE, Farb A, Weber KT. Pathologic remodeling of the myocardium in a weightlifter taking anabolic steroids. Blood pressure. 1993;2(3):213–6. - PubMed
-
- Marsh JD, Lehmann MH, Ritchie RH, Gwathmey JK, Green GE, Schiebinger RJ. Androgen receptors mediate hypertrophy in cardiac myocytes. Circulation. 1998;98(3): 256–261. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
