

Mucopolysaccharidosis IVA and glycosaminoglycans
- PMID: 27979613
- PMCID: PMC5293636
- DOI: 10.1016/j.ymgme.2016.11.007
Mucopolysaccharidosis IVA and glycosaminoglycans
Abstract
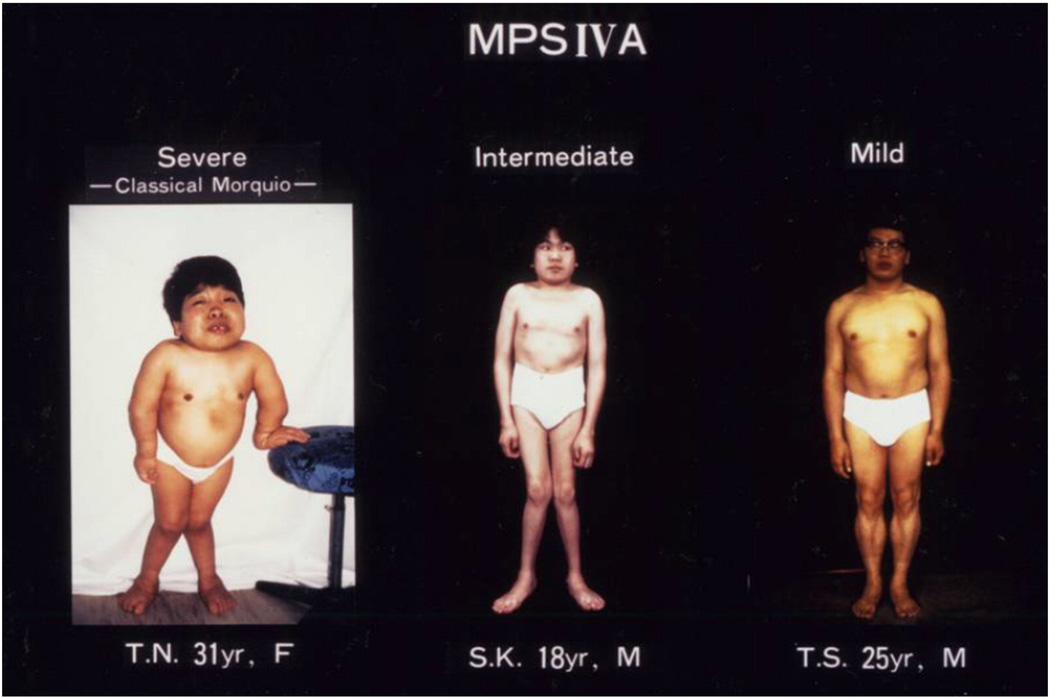
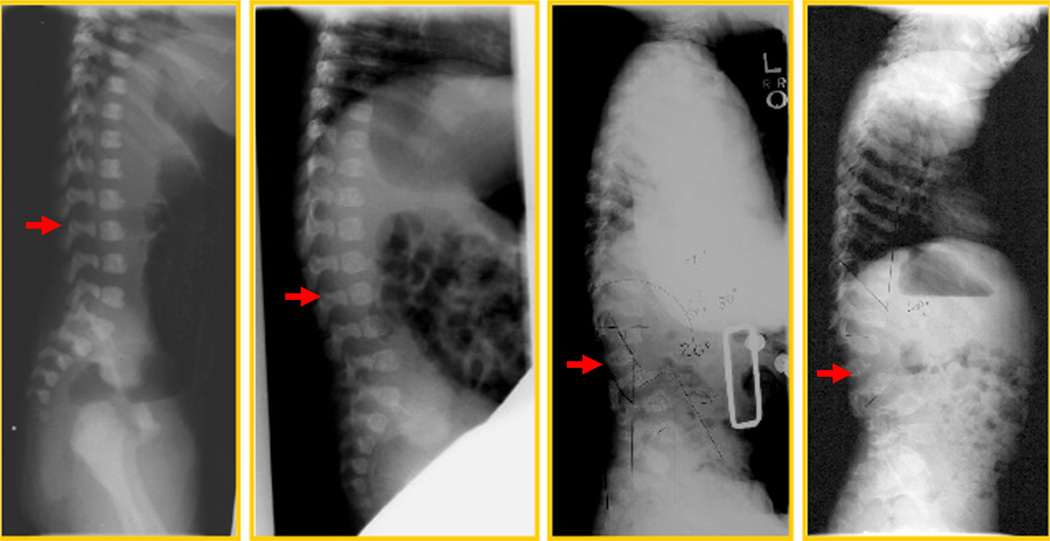
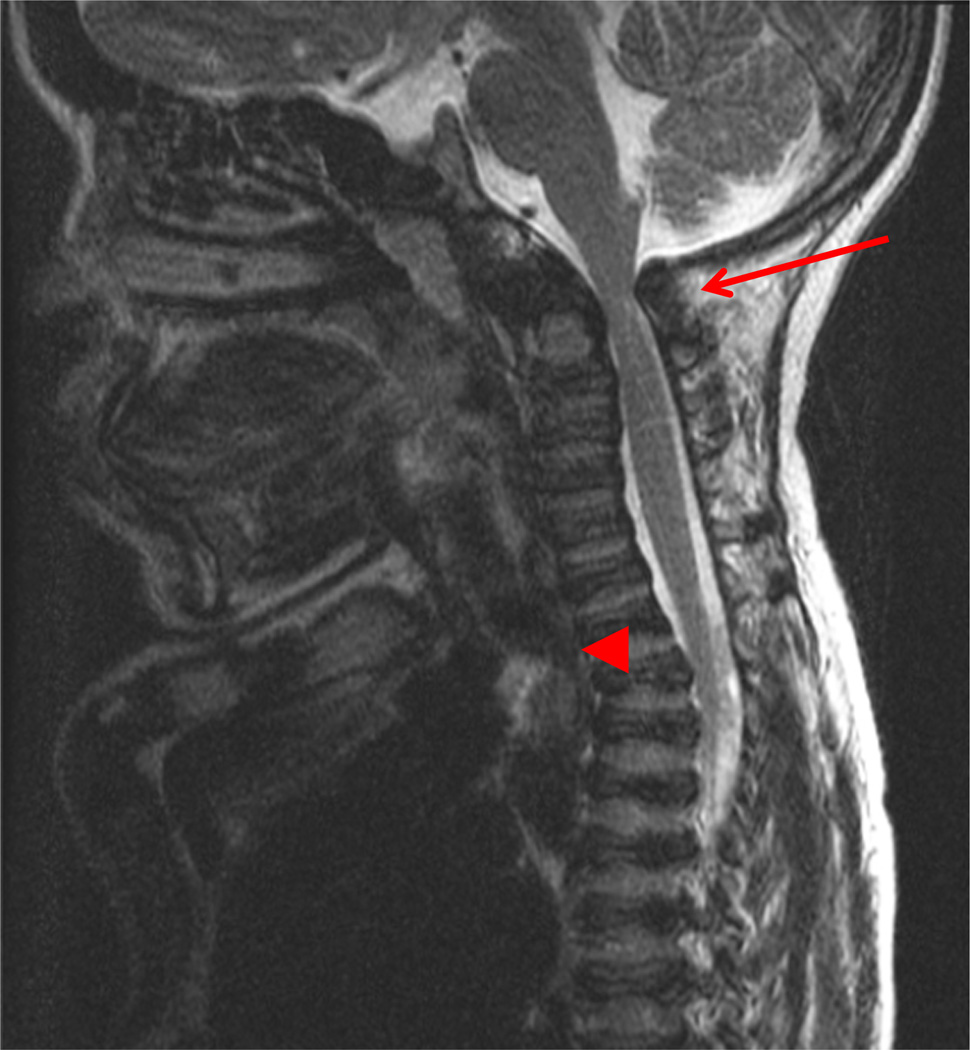
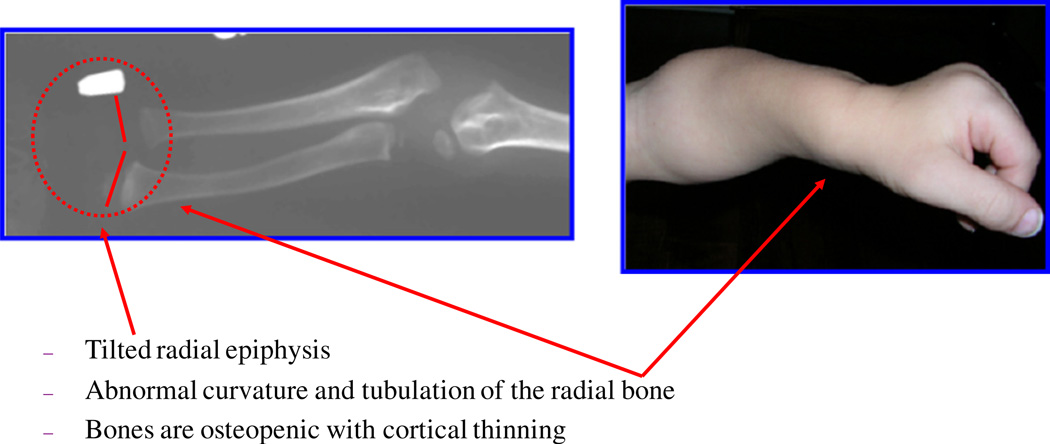
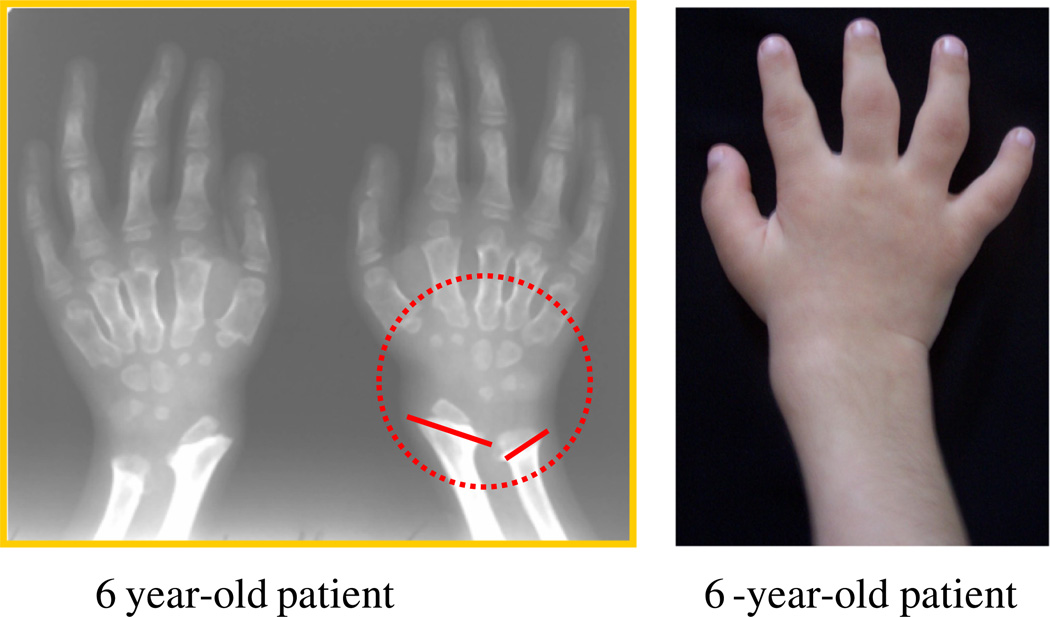
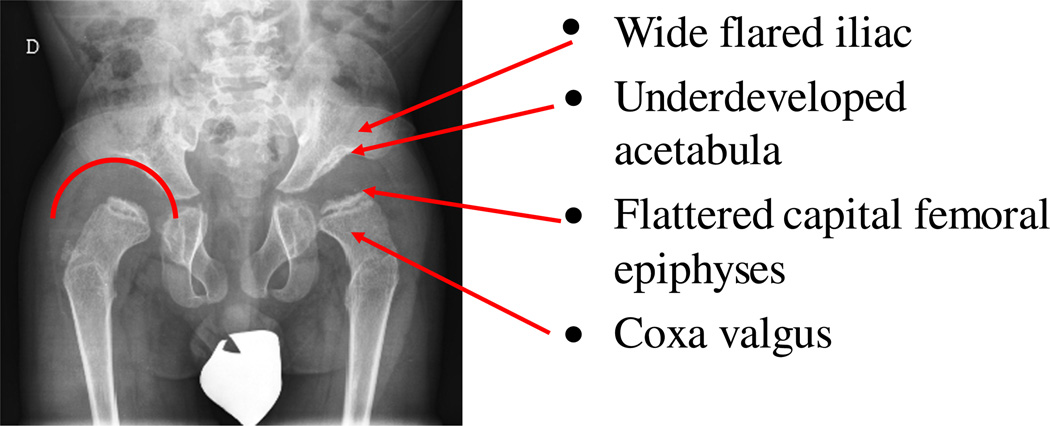
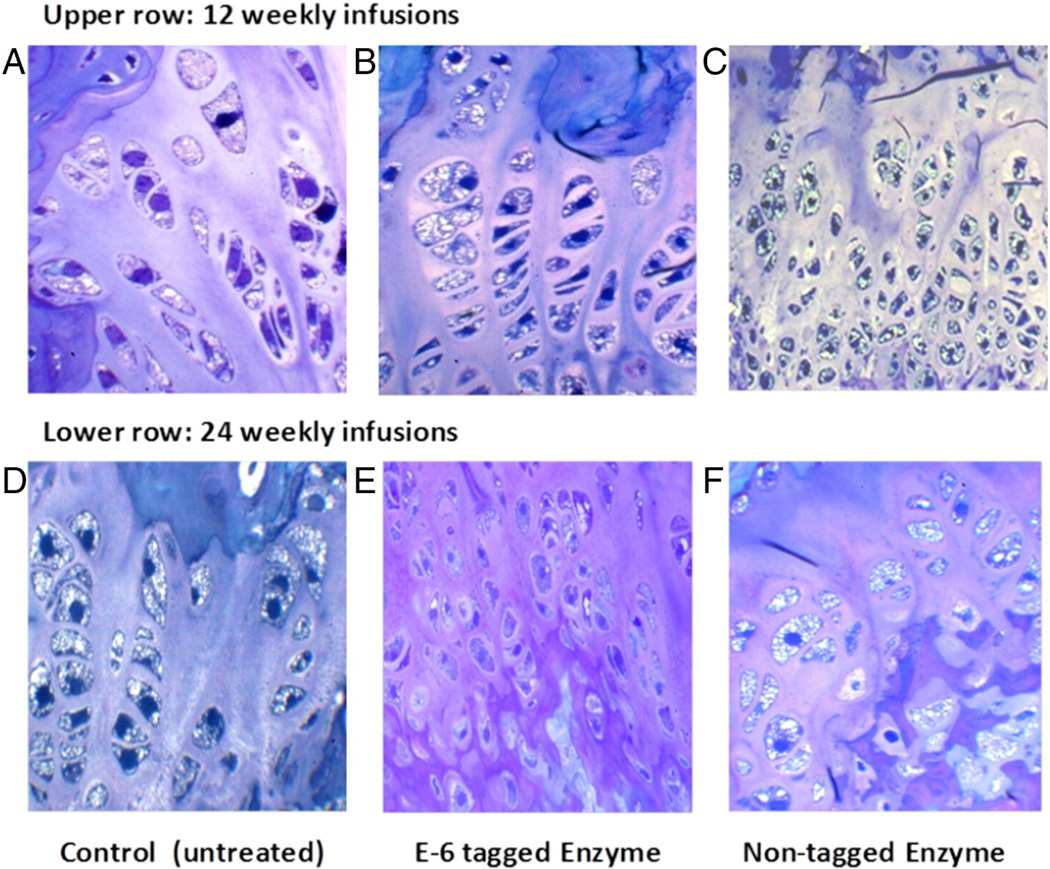
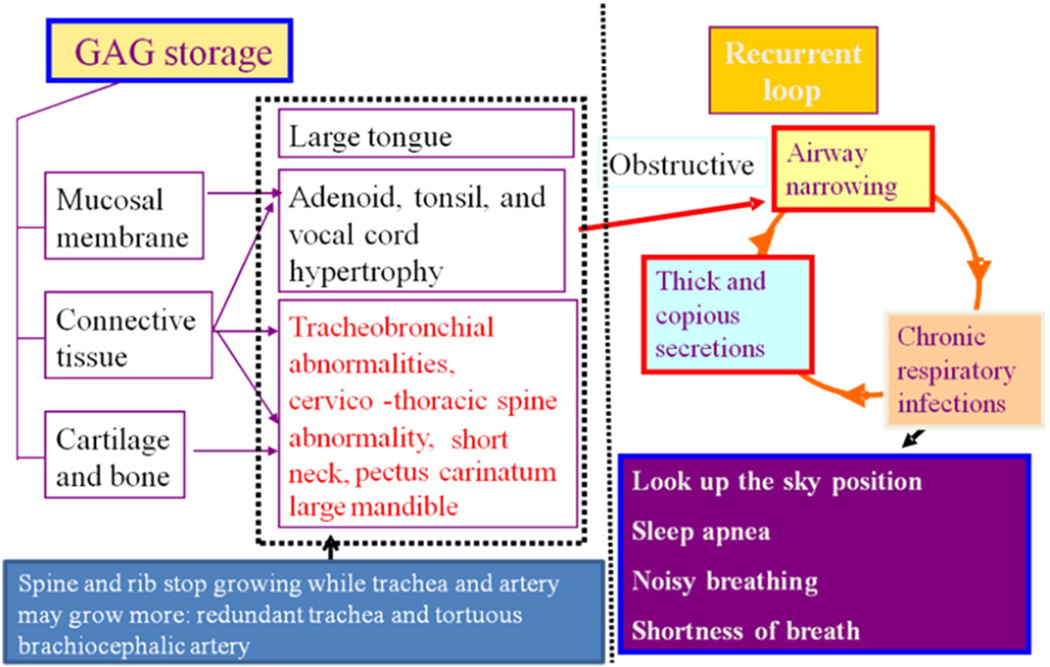
Mucopolysaccharidosis IVA (MPS IVA; Morquio A: OMIM 253000) is a lysosomal storage disease with an autosomal recessive trait caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase. Deficiency of this enzyme leads to accumulation of specific glycosaminoglycans (GAGs): chondroitin-6-sulfate (C6S) and keratan sulfate (KS). C6S and KS are mainly produced in the cartilage. Therefore, the undegraded substrates are stored primarily in cartilage and in its extracellular matrix (ECM), leading to a direct impact on cartilage and bone development, and successive systemic skeletal dysplasia. Chondrogenesis, the earliest phase of skeletal formation, is maintained by cellular interactions with the ECM, growth and differentiation factors, signaling pathways, and transcription factors in a temporal-spatial manner. In patients with MPS IVA, the cartilage is disrupted at birth as a consequence of abnormal chondrogenesis and/or endochondral ossification. The unique skeletal features are distinguished by a disproportional short stature, odontoid hypoplasia, spinal cord compression, tracheal obstruction, pectus carinatum, kyphoscoliosis, platyspondyly, coxa valga, genu valgum, waddling gait, and laxity of joints. In spite of many descriptions of these unique clinical features, delay of diagnosis still happens. The pathogenesis and treatment of systemic skeletal dysplasia in MPS IVA remains an unmet challenge. In this review article, we comprehensively describe historical aspect, property of GAGs, diagnosis, screening, pathogenesis, and current and future therapies of MPS IVA.
Keywords: Chondroitin-6-sulfate; Keratan sulfate; Mucopolysaccharidosis IVA; N-acetylgalactosamine-6-sulfate sulfatase; Skeletal dysplasia.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interestAll the authors contributed to the Review Article and had no conflict of interest with any other party.Shaukat Khan, Carlos J. Alméciga-Díaz, Kazuki Sawamoto, William G. Mackenzie, Mary C. Theroux, Christian Pizarro, Robert W. Mason, Tadao Orii, and Shunji Tomatsu declare that they have no conflict of interests.
Figures
References
-
- Morquio L. Sur une forme de dystrophic osseuse familiale. Arch. Med. Infant. 1929;32:129–135.
-
- JF B. Chondro-osteo-dystrophy. Roentgenographic and clinical features of a child with dislocation of vertebrae. Am. J. Surg. 1929;7:404–410. - PubMed
-
- Pedrini V, Lennzi L, Zambotti V. Isolation and identification of keratosulphate in urine of patients affected by Morquio-Ullrich disease. Proc. Soc. Exp. Biol. Med. 1962;110:847–849. - PubMed
-
- McKusick VA, Kaplan D, Wise D, Hanley WB, Suddarth SB, Sevick ME, Maumanee AE. The genetic mucopolysaccharidoses. Medicine. 1965;44:445–483. - PubMed
-
- Orii T, Minami R, Chiba T, Yamaguchi M, Tsugawa S, Nakao T, Horino K, Sakuma K. Study on Morquio syndrome bone metabolism (in Japanese) 1971;5:72–78.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
