

Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study
- PMID: 27979989
- PMCID: PMC5407735
- DOI: 10.1681/ASN.2016090964
Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study
Abstract
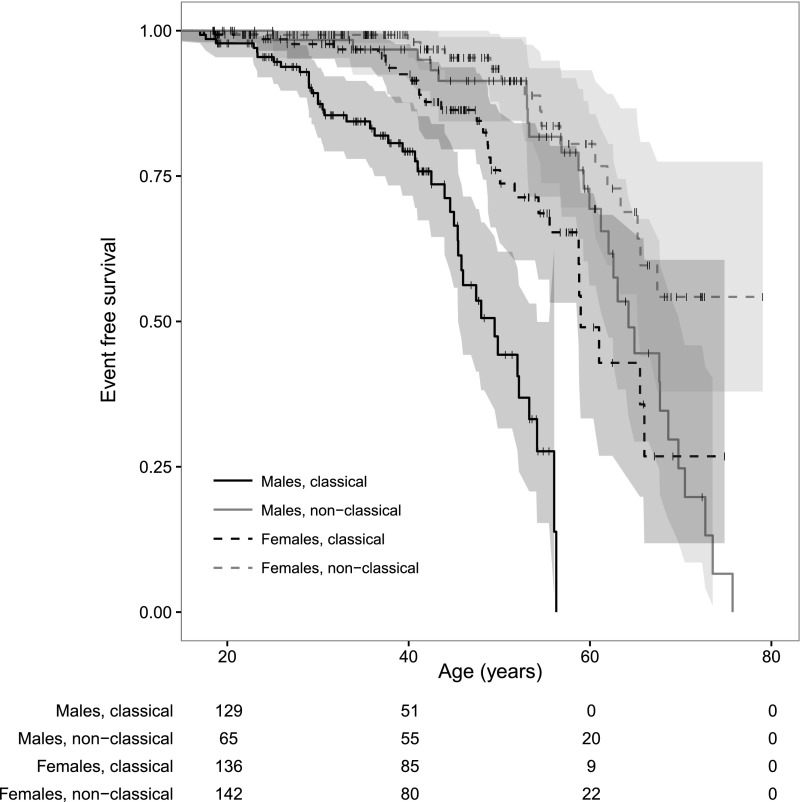
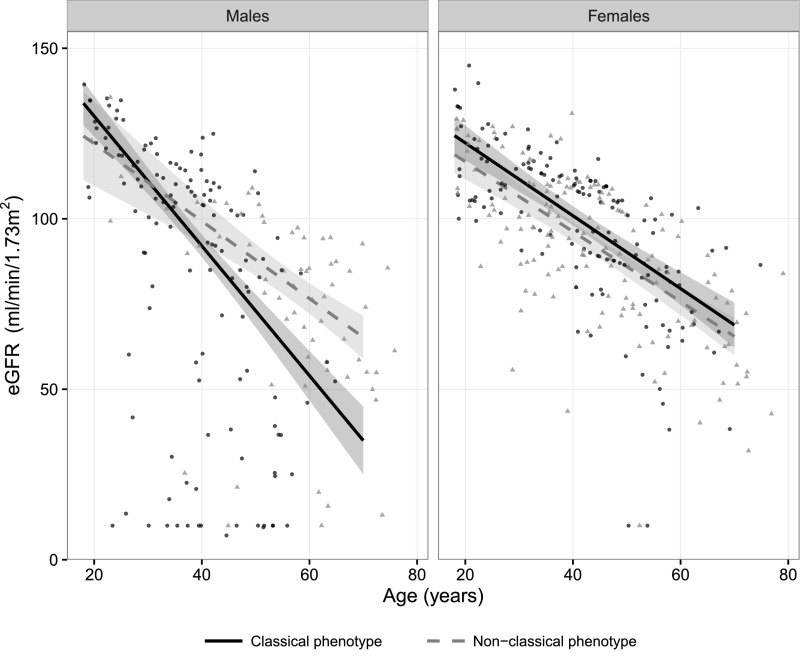
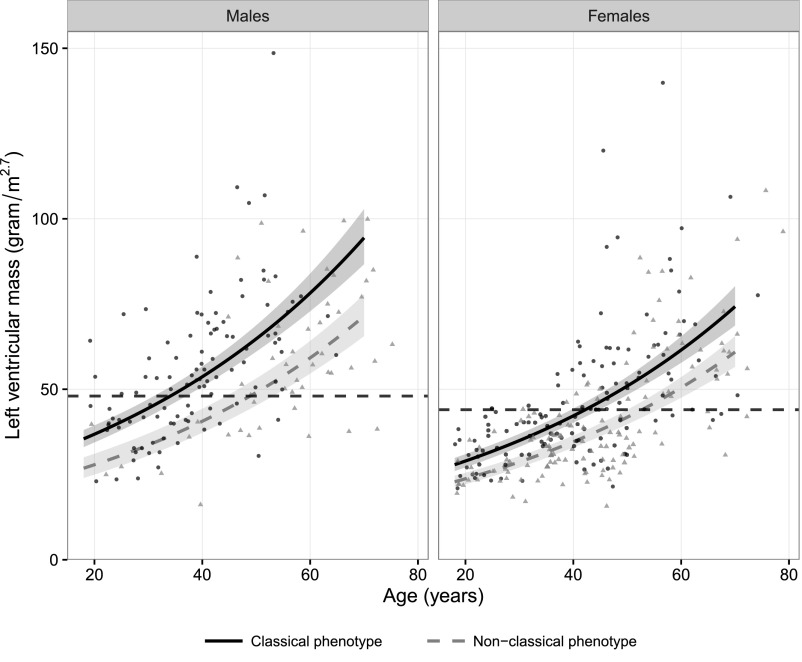
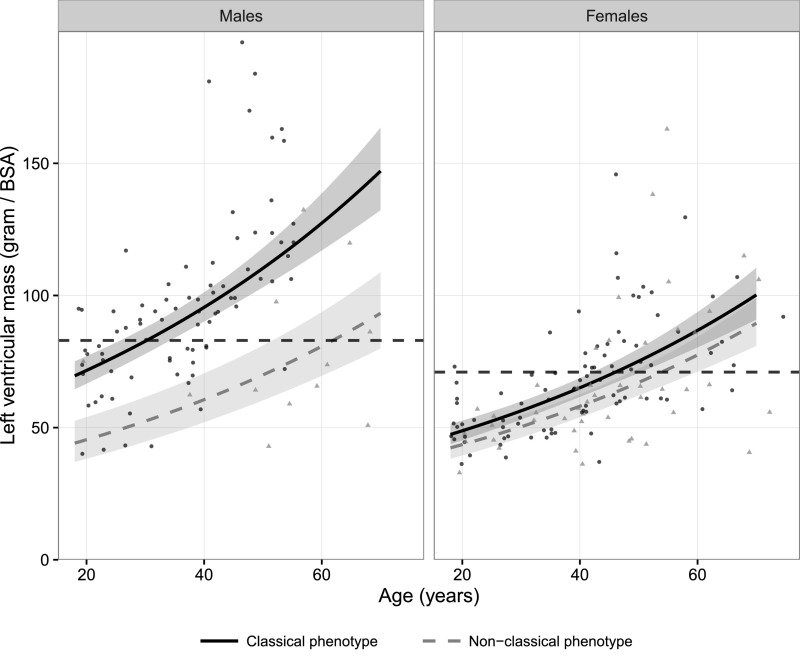
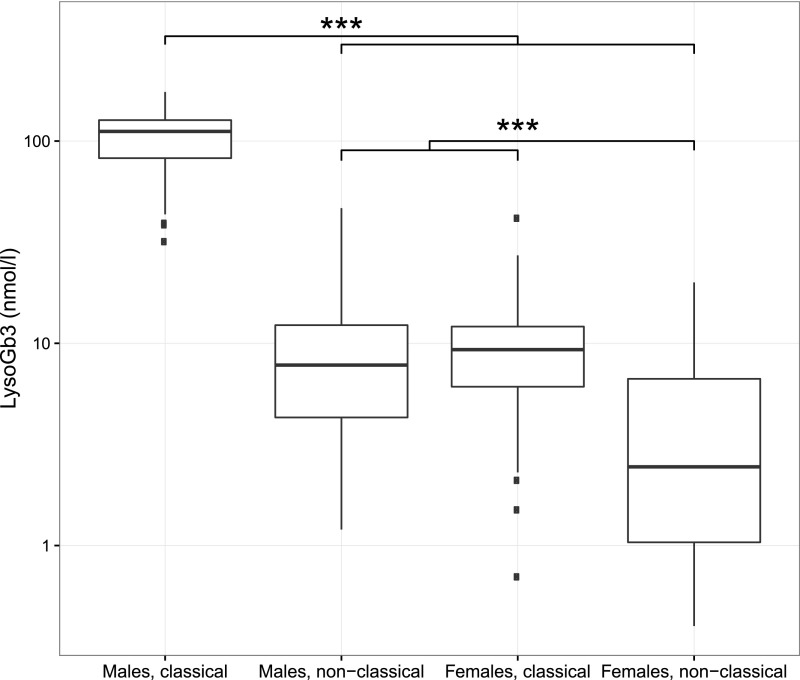
Fabry disease leads to renal, cardiac, and cerebrovascular manifestations. Phenotypic differences between classically and nonclassically affected patients are evident, but there are few data on the natural course of classical and nonclassical disease in men and women. To describe the natural course of Fabry disease stratified by sex and phenotype, we retrospectively assessed event-free survival from birth to the first clinical visit (before enzyme replacement therapy) in 499 adult patients (mean age 43 years old; 41% men; 57% with the classical phenotype) from three international centers of excellence. We classified patients by phenotype on the basis of characteristic symptoms and enzyme activity. Men and women with classical Fabry disease had higher event rate than did those with nonclassical disease (hazard ratio for men, 5.63, 95% confidence interval, 3.17 to 10.00; P<0.001; hazard ratio for women, 2.88, 95% confidence interval, 1.54 to 5.40; P<0.001). Furthermore, men with classical Fabry disease had lower eGFR, higher left ventricular mass, and higher plasma globotriaosylsphingosine concentrations than men with nonclassical Fabry disease or women with either phenotype (P<0.001). In conclusion, before treatment with enzyme replacement therapy, men with classical Fabry disease had a history of more events than men with nonclassical disease or women with either phenotype; women with classical Fabry disease were more likely to develop complications than women with nonclassical disease. These data may support the development of new guidelines for the monitoring and treatment of Fabry disease and studies on the effects of intervention in subgroups of patients.
Keywords: Fabry-s disease; alpha galactosidase A; natural disease course; natural history; phenotype.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- Kint JA: Fabry’s disease: Alpha-galactosidase deficiency. Science 167: 1268–1269, 1970 - PubMed
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L: Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med 276: 1163–1167, 1967 - PubMed
-
- Desnick RJ, Ioannou YA, Eng CM: A-galactosidase a deficiency: Fabry disease. In: The Online Metabolic and Molecular Bases of Inherited Diseases, edited by Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, New York, McGraw-Hill, 2014; Available at http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62644837. Accessed November 24, 2016
-
- Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ: Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet 52: 262–268, 2015 - PubMed
-
- van der Tol L, Smid BE, Poorthuis BJ, Biegstraaten M, Deprez RH, Linthorst GE, Hollak CE: A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J Med Genet 51: 1–9, 2014 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
