

Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort
- PMID: 27980096
- PMCID: PMC5389578
- DOI: 10.1093/nar/gkw1237
Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort
Abstract
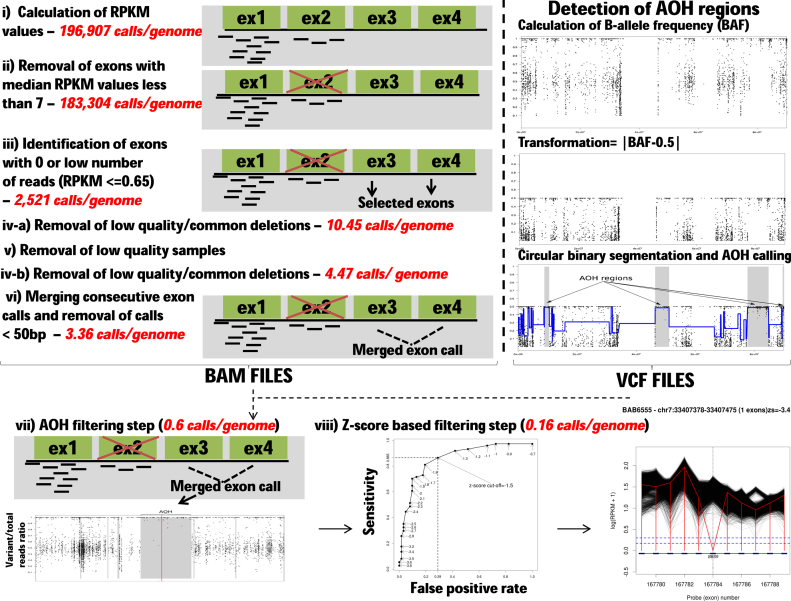
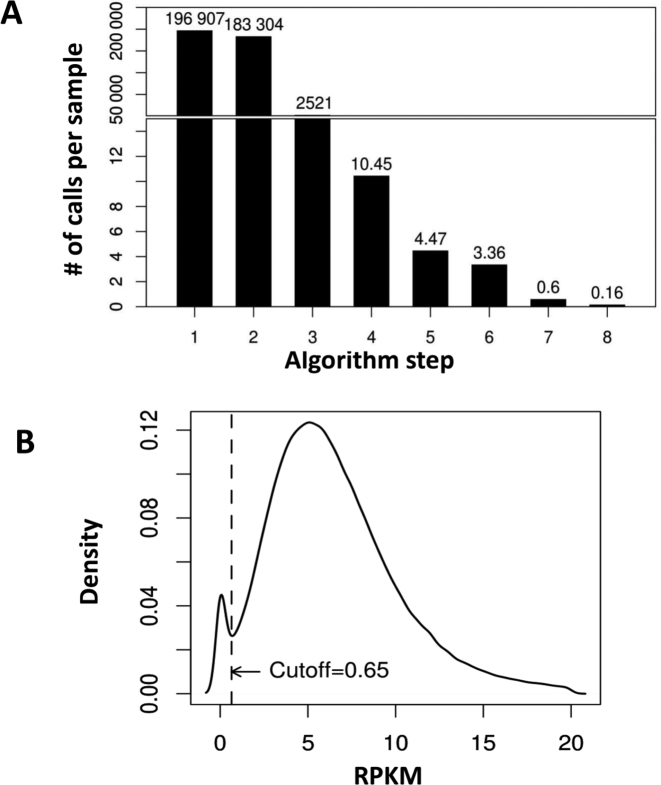
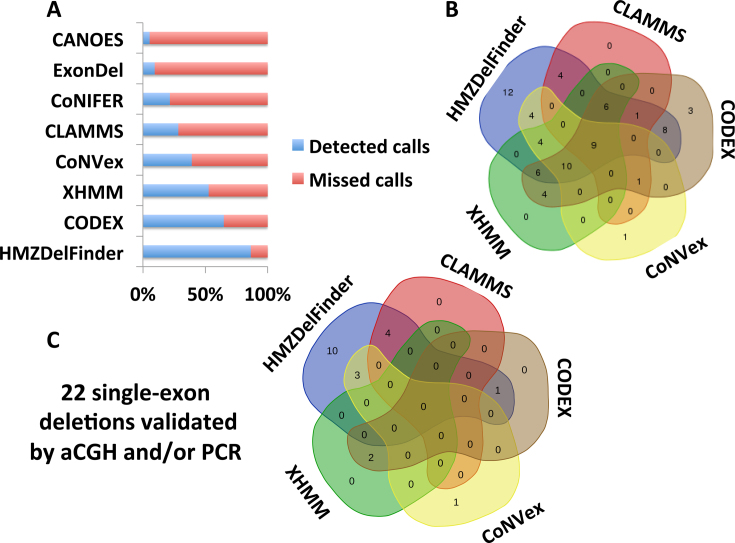
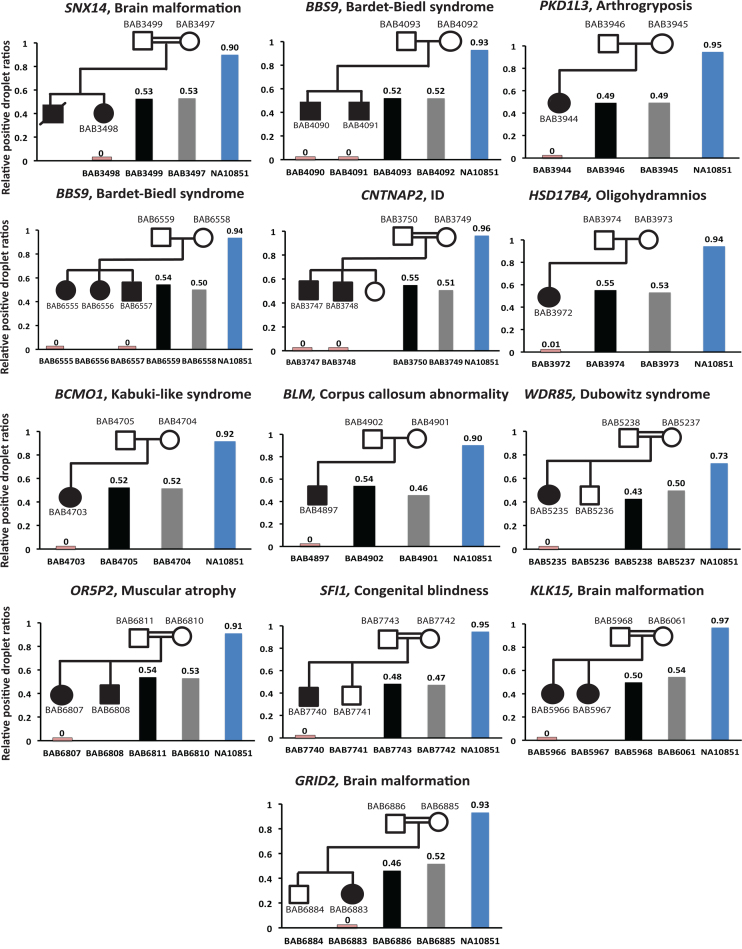
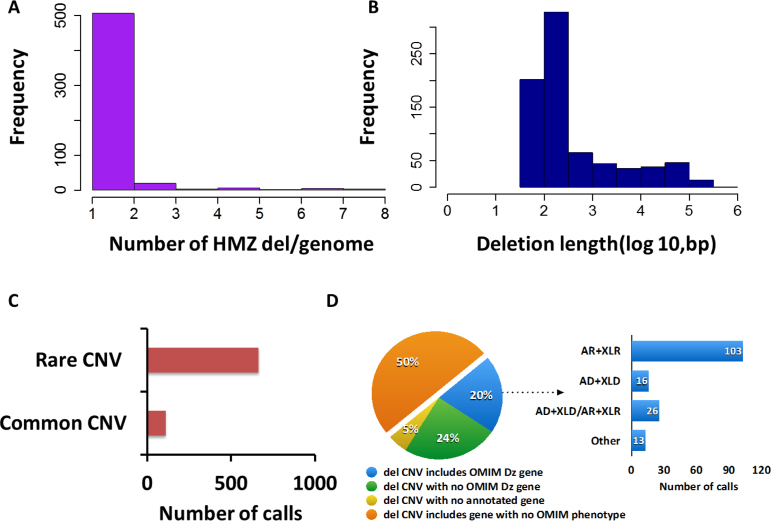
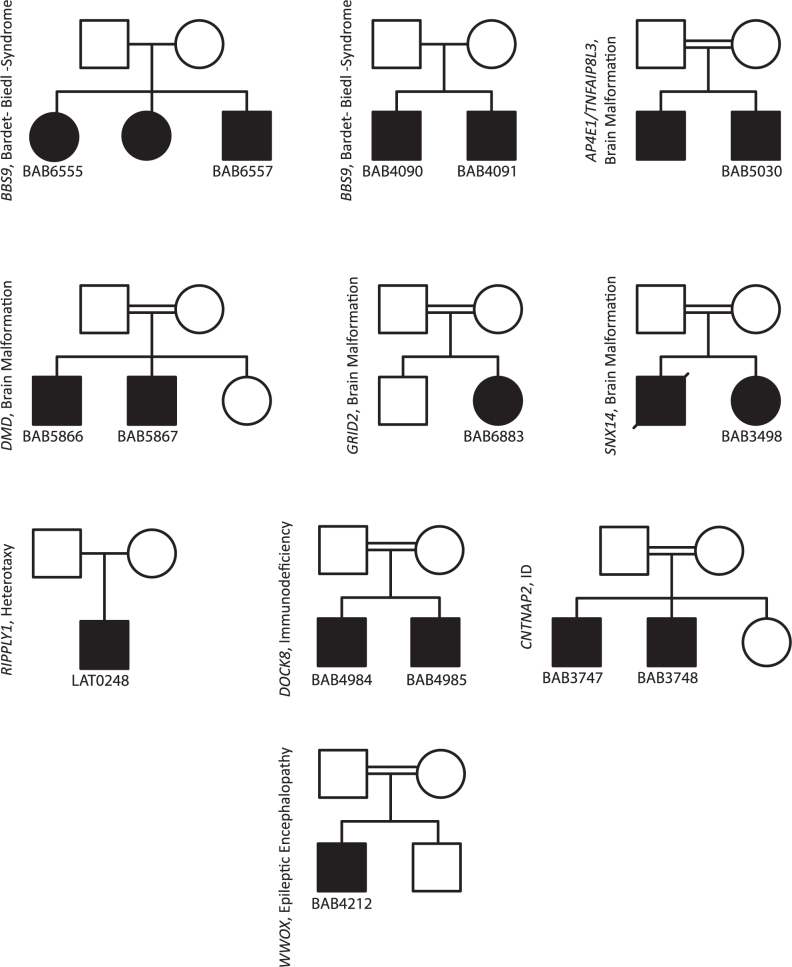
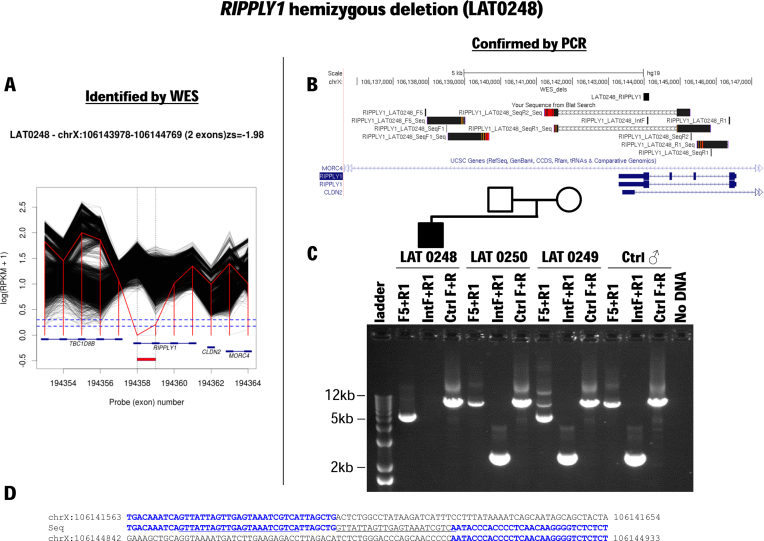
We developed an algorithm, HMZDelFinder, that uses whole exome sequencing (WES) data to identify rare and intragenic homozygous and hemizygous (HMZ) deletions that may represent complete loss-of-function of the indicated gene. HMZDelFinder was applied to 4866 samples in the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) cohort and detected 773 HMZ deletion calls (567 homozygous or 206 hemizygous) with an estimated sensitivity of 86.5% (82% for single-exonic and 88% for multi-exonic calls) and precision of 78% (53% single-exonic and 96% for multi-exonic calls). Out of 773 HMZDelFinder-detected deletion calls, 82 were subjected to array comparative genomic hybridization (aCGH) and/or breakpoint PCR and 64 were confirmed. These include 18 single-exon deletions out of which 8 were exclusively detected by HMZDelFinder and not by any of seven other CNV detection tools examined. Further investigation of the 64 validated deletion calls revealed at least 15 pathogenic HMZ deletions. Of those, 7 accounted for 17-50% of pathogenic CNVs in different disease cohorts where 7.1-11% of the molecular diagnosis solved rate was attributed to CNVs. In summary, we present an algorithm to detect rare, intragenic, single-exon deletion CNVs using WES data; this tool can be useful for disease gene discovery efforts and clinical WES analyses.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Stankiewicz P., Lupski J.R.. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010; 61:437–455. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
