

"Gap hunting" to characterize clustered probe signals in Illumina methylation array data
- PMID: 27980682
- PMCID: PMC5142147
- DOI: 10.1186/s13072-016-0107-z
"Gap hunting" to characterize clustered probe signals in Illumina methylation array data
Abstract
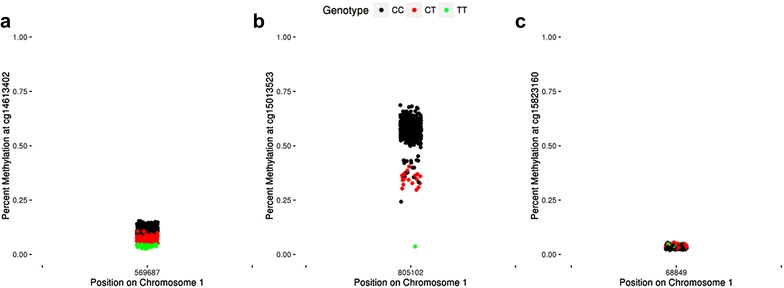
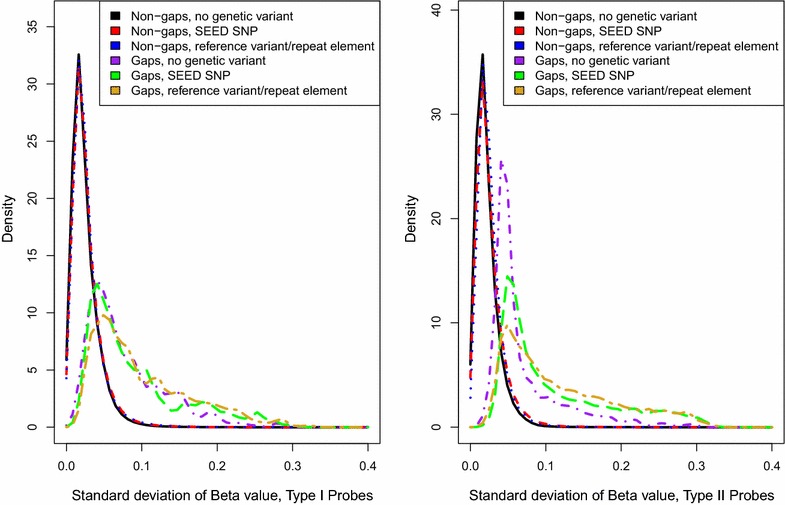
Background: The Illumina 450k array has been widely used in epigenetic association studies. Current quality-control (QC) pipelines typically remove certain sets of probes, such as those containing a SNP or with multiple mapping locations. An additional set of potentially problematic probes are those with DNA methylation distributions characterized by two or more distinct clusters separated by gaps. Data-driven identification of such probes may offer additional insights for downstream analyses.
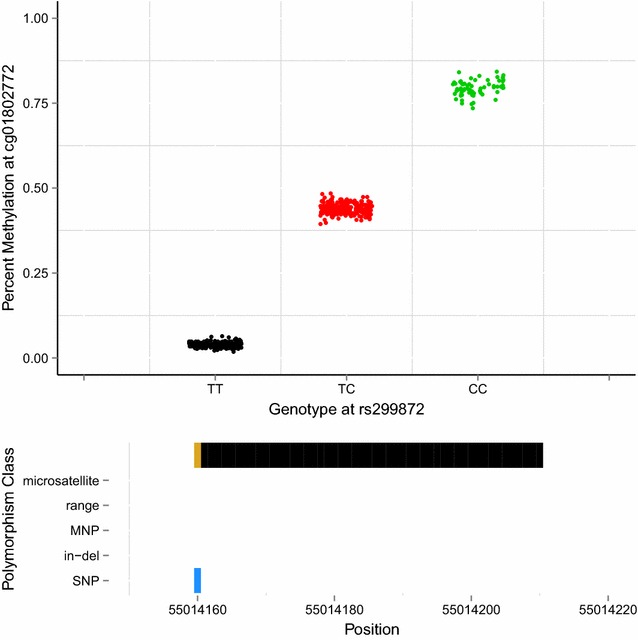
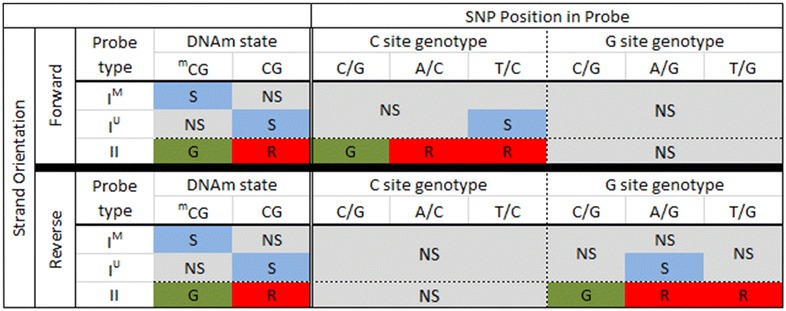
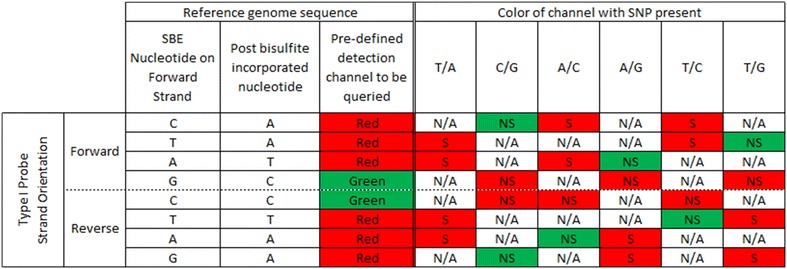
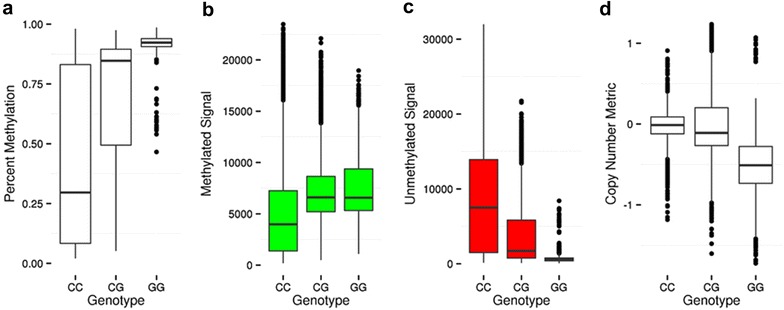
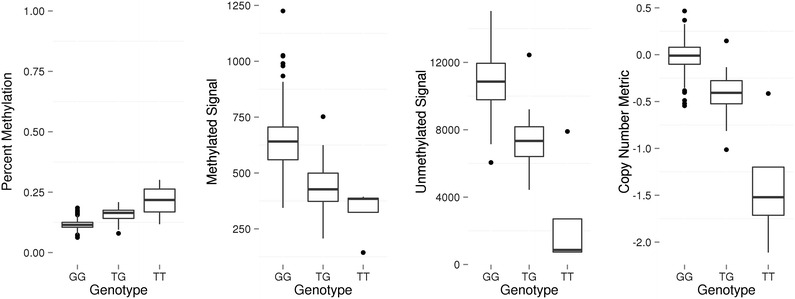
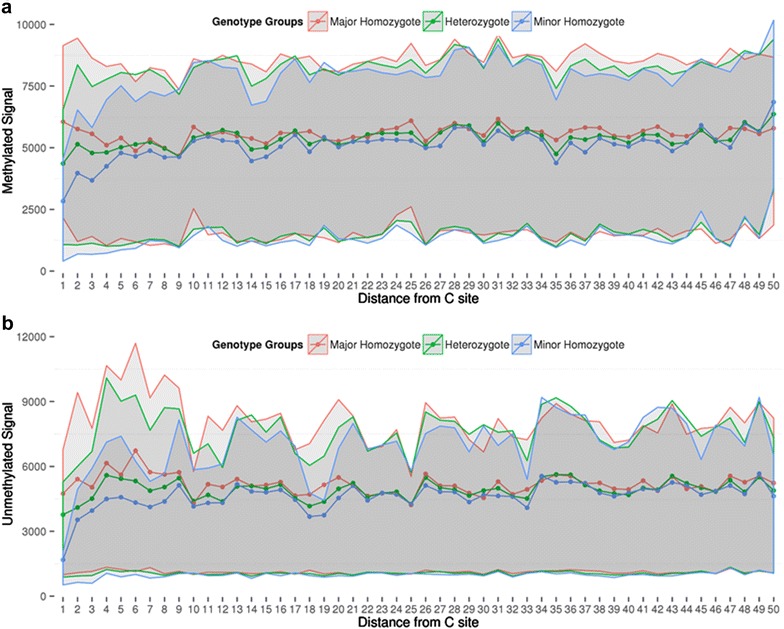
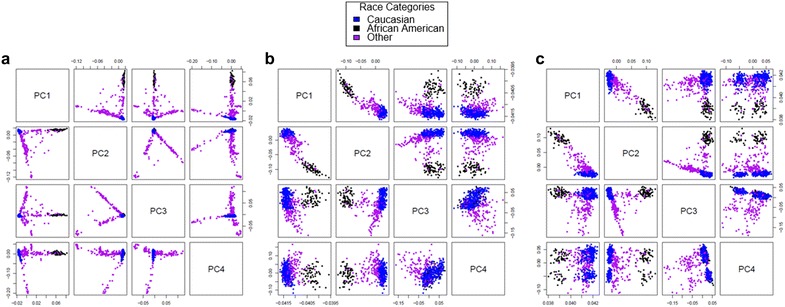
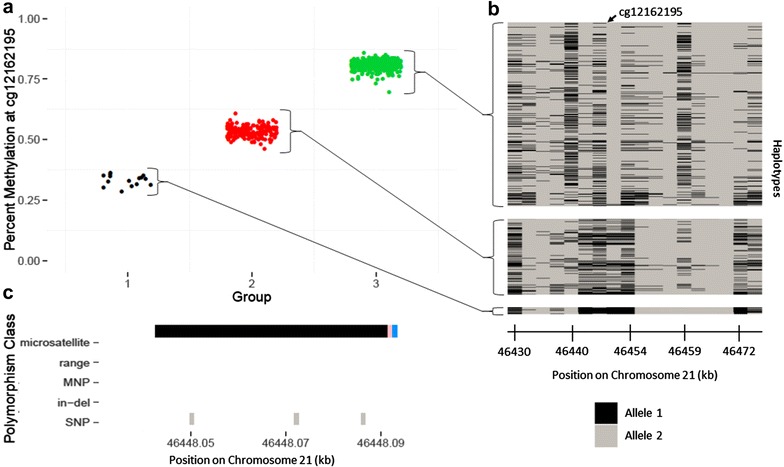
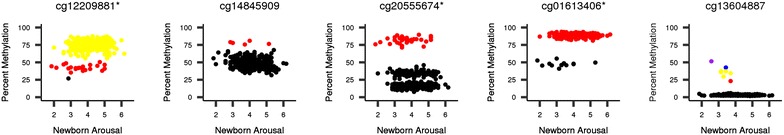
Results: We developed a procedure, termed "gap hunting," to identify probes showing clustered distributions. Among 590 peripheral blood samples from the Study to Explore Early Development, we identified 11,007 "gap probes." The vast majority (9199) are likely attributed to an underlying SNP(s) or other variant in the probe, although SNP-affected probes exist that do not produce a gap signals. Specific factors predict which SNPs lead to gap signals, including type of nucleotide change, probe type, DNA strand, and overall methylation state. These expected effects are demonstrated in paired genotype and 450k data on the same samples. Gap probes can also serve as a surrogate for the local genetic sequence on a haplotype scale and can be used to adjust for population stratification.
Conclusions: The characteristics of gap probes reflect potentially informative biology. QC pipelines may benefit from an efficient data-driven approach that "flags" gap probes, rather than filtering such probes, followed by careful interpretation of downstream association analyses. Our results should translate directly to the recently released Illumina EPIC array given the similar chemistry and content design.
Keywords: 450k Array; Epigenome-wide association studies; Gap hunting; Illumina HumanMethylation450 BeadChip; Polymorphic CpG; SNP.
Figures
References
-
- Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, Shchetynsky K, Scheynius A, Kere J, Alfredsson L, Klareskog L, Ekström TJ, Feinberg AP. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–147. doi: 10.1038/nbt.2487. - DOI - PMC - PubMed
-
- Ladd-Acosta C, Shu C, Lee BK, Gidaya N, Singer A, Schieve LA, Schendel DE, Jones N, Daniels JL, Windham GC, Newschaffer CJ, Croen LA, Feinberg AP, Daniele Fallin M. Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ Res. 2016; 144(Pt A):139–148. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
