

Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens
- PMID: 27984732
- PMCID: PMC5181115
- DOI: 10.1016/j.cell.2016.11.038
Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens
Abstract
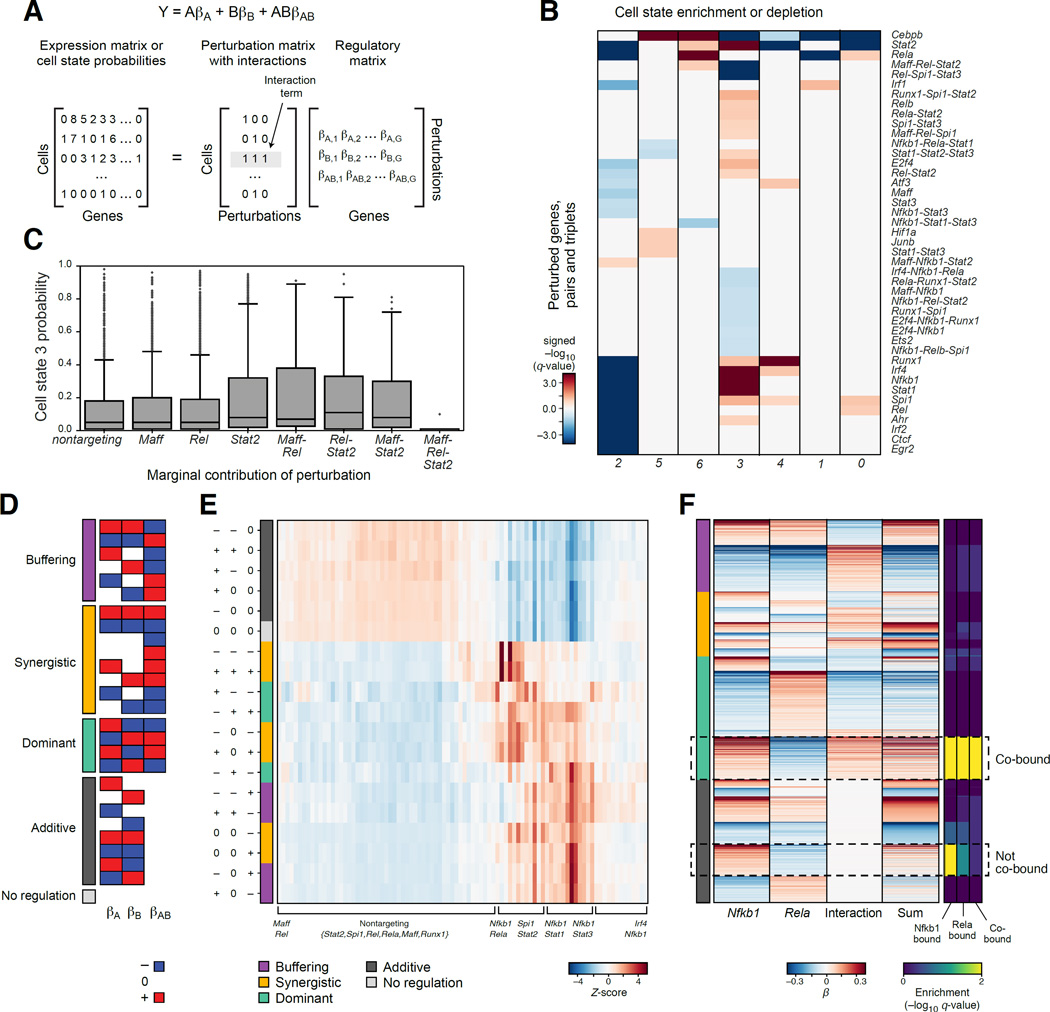
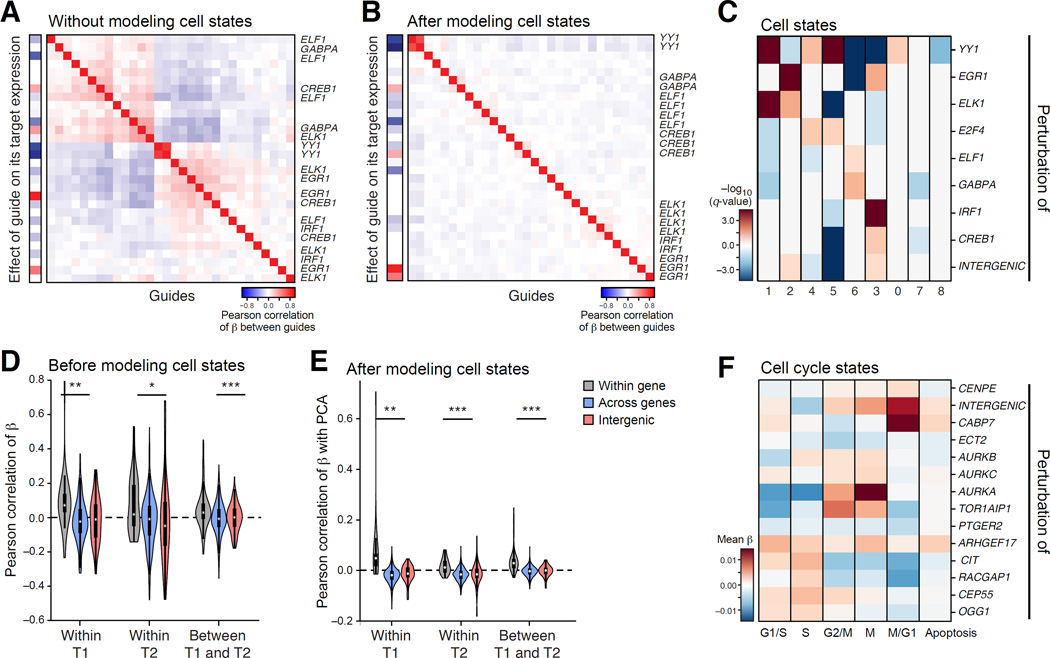
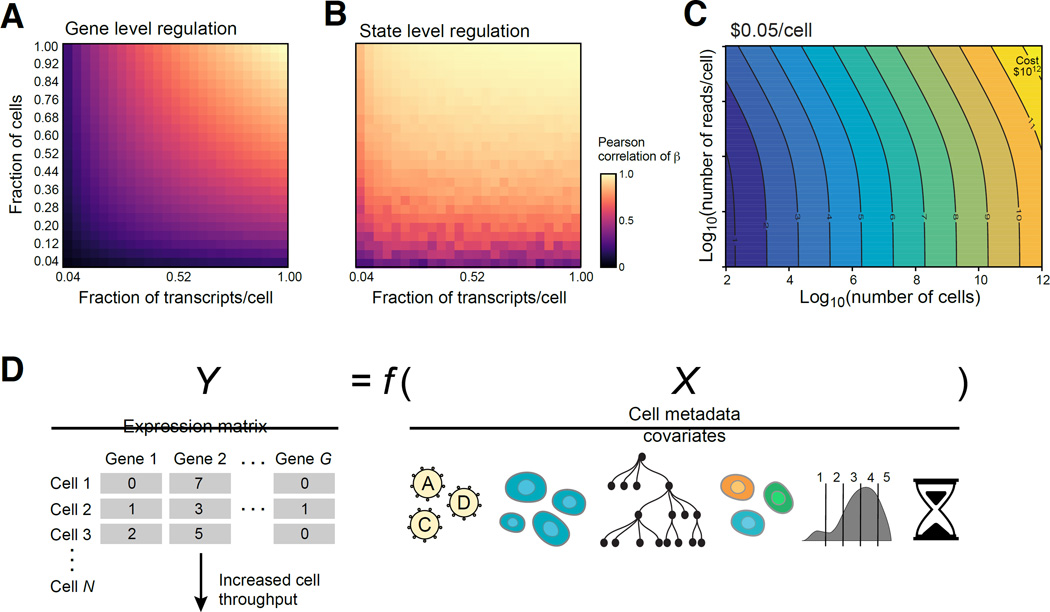
Genetic screens help infer gene function in mammalian cells, but it has remained difficult to assay complex phenotypes-such as transcriptional profiles-at scale. Here, we develop Perturb-seq, combining single-cell RNA sequencing (RNA-seq) and clustered regularly interspaced short palindromic repeats (CRISPR)-based perturbations to perform many such assays in a pool. We demonstrate Perturb-seq by analyzing 200,000 cells in immune cells and cell lines, focusing on transcription factors regulating the response of dendritic cells to lipopolysaccharide (LPS). Perturb-seq accurately identifies individual gene targets, gene signatures, and cell states affected by individual perturbations and their genetic interactions. We posit new functions for regulators of differentiation, the anti-viral response, and mitochondrial function during immune activation. By decomposing many high content measurements into the effects of perturbations, their interactions, and diverse cell metadata, Perturb-seq dramatically increases the scope of pooled genomic assays.
Keywords: CRISPR; epistasis; genetic interactions; pooled screen; single-cell RNA-seq.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
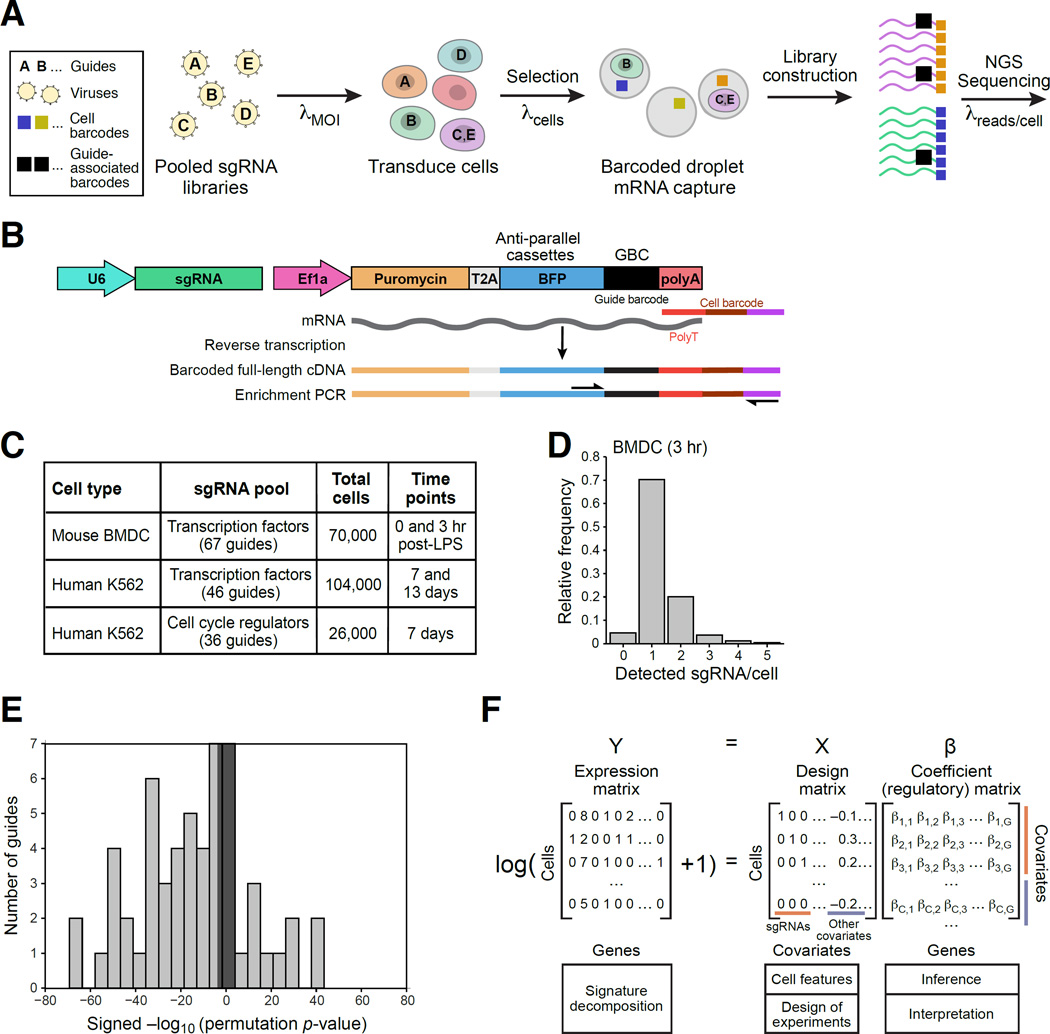
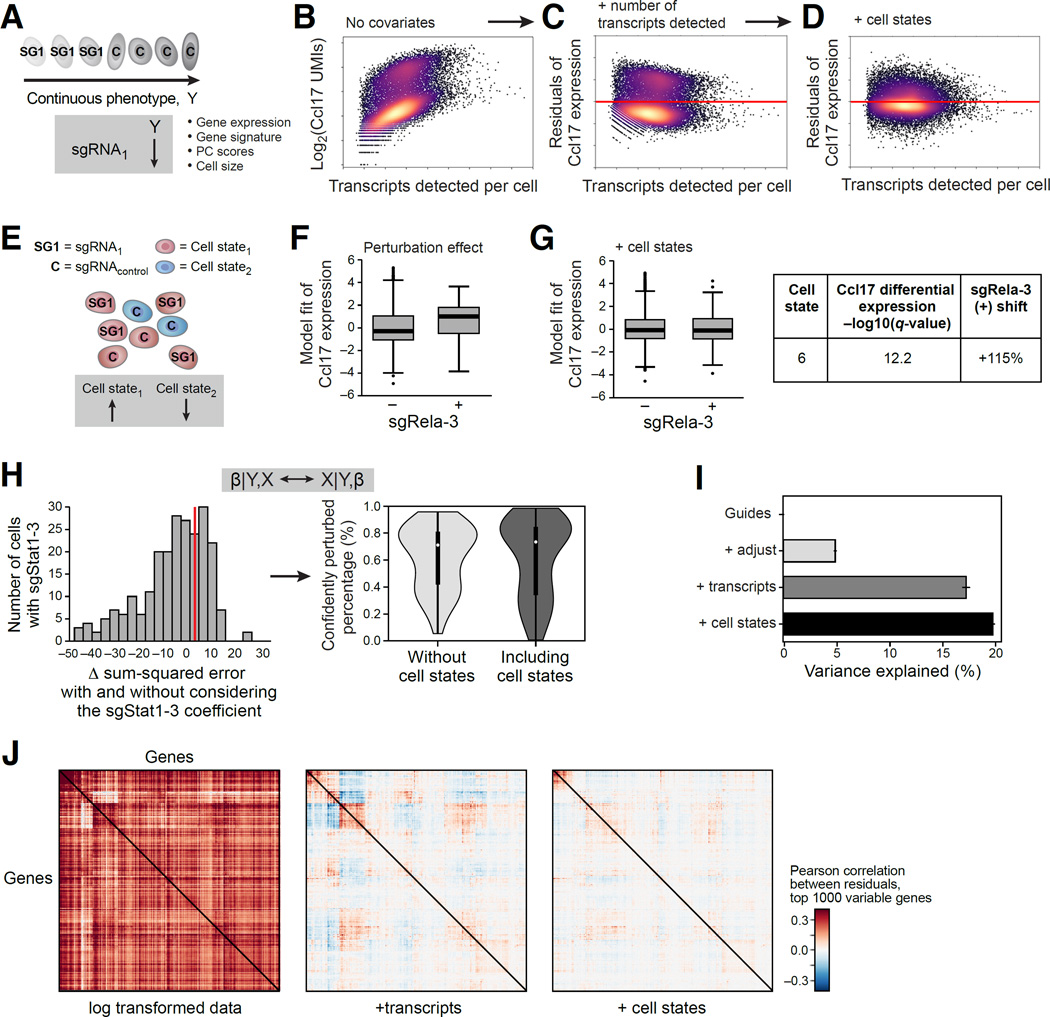
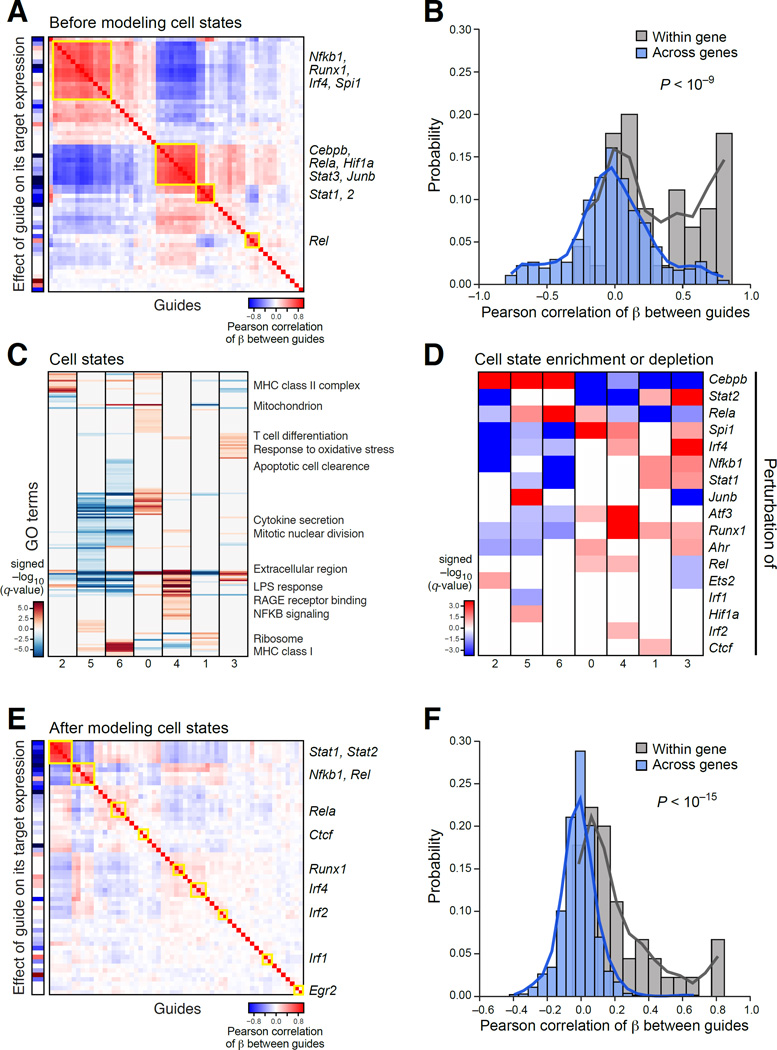
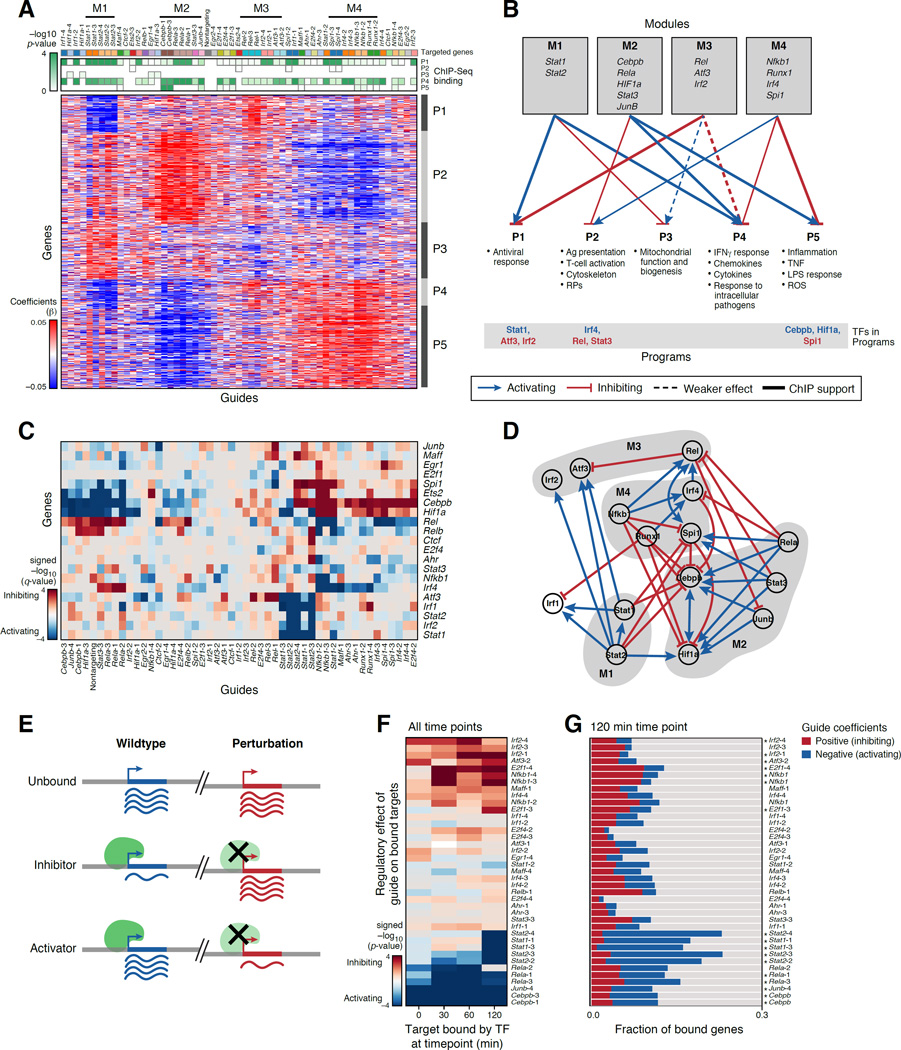
Comment in
-
Genetic screens: Combining CRISPR perturbations and RNA-seq.Nat Rev Genet. 2017 Jan 18;18(2):67. doi: 10.1038/nrg.2016.171. Nat Rev Genet. 2017. PMID: 28096529 No abstract available.
-
Genetic screening enters the single-cell era.Nat Methods. 2017 Feb 28;14(3):237-238. doi: 10.1038/nmeth.4196. Nat Methods. 2017. PMID: 28245215 No abstract available.
-
Single-minded CRISPR screening.Nat Biotechnol. 2017 Apr 11;35(4):339-340. doi: 10.1038/nbt.3849. Nat Biotechnol. 2017. PMID: 28398325 No abstract available.
References
-
- Beerenwinkel N, Pachter L, Sturmfels B. Epistasis and Shapes of Fitness Landscapes. Stat. Sin. 2007;17:1317–1342.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
