

A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response
- PMID: 27984733
- PMCID: PMC5315571
- DOI: 10.1016/j.cell.2016.11.048
A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response
Abstract
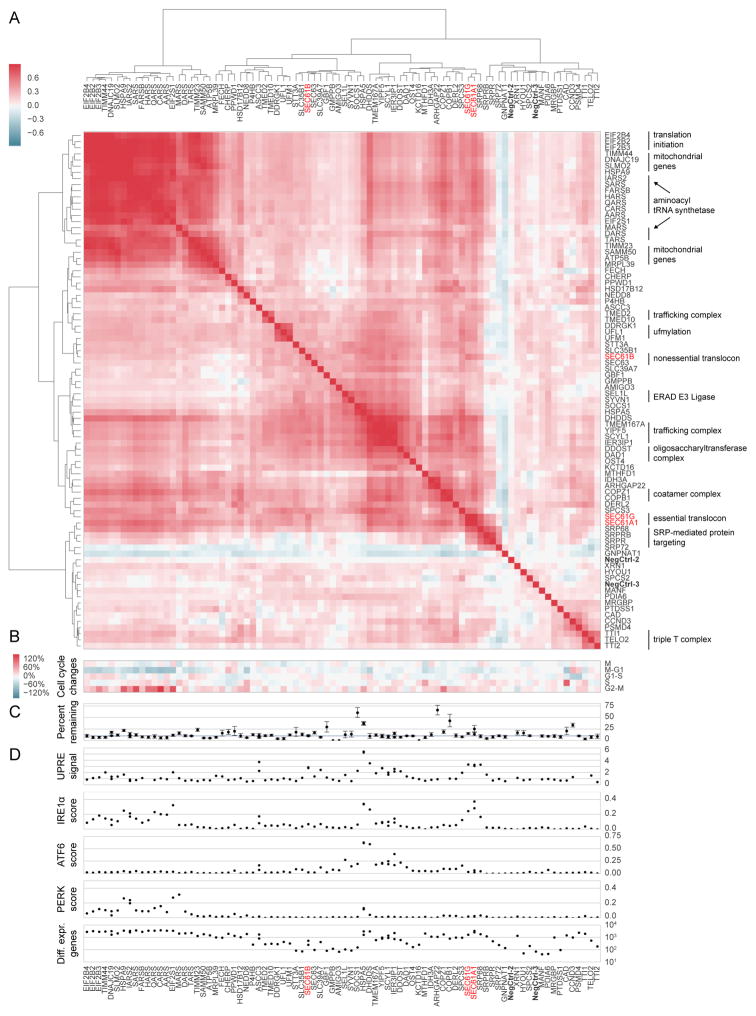
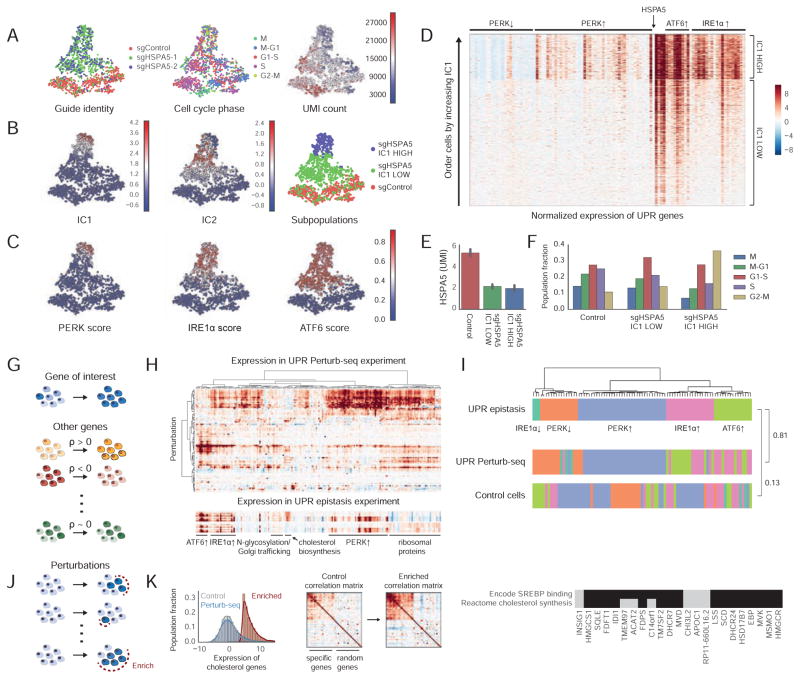
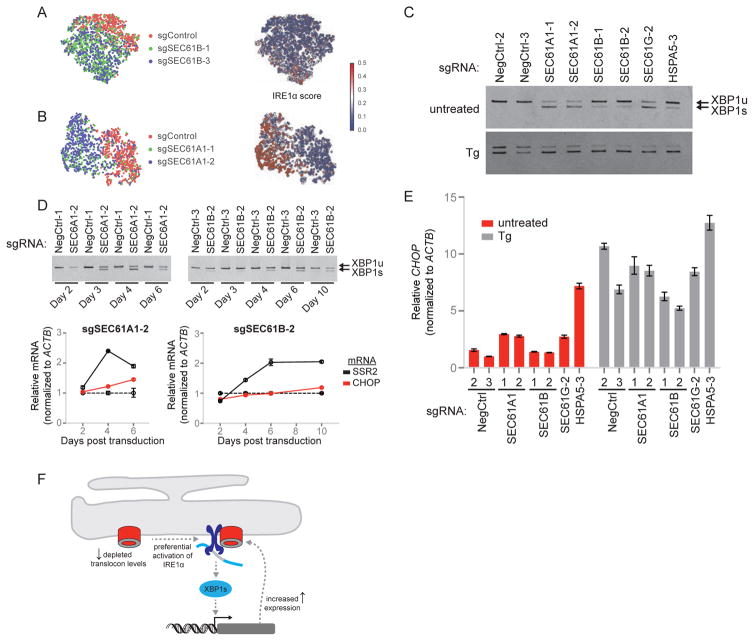
Functional genomics efforts face tradeoffs between number of perturbations examined and complexity of phenotypes measured. We bridge this gap with Perturb-seq, which combines droplet-based single-cell RNA-seq with a strategy for barcoding CRISPR-mediated perturbations, allowing many perturbations to be profiled in pooled format. We applied Perturb-seq to dissect the mammalian unfolded protein response (UPR) using single and combinatorial CRISPR perturbations. Two genome-scale CRISPR interference (CRISPRi) screens identified genes whose repression perturbs ER homeostasis. Subjecting ∼100 hits to Perturb-seq enabled high-precision functional clustering of genes. Single-cell analyses decoupled the three UPR branches, revealed bifurcated UPR branch activation among cells subject to the same perturbation, and uncovered differential activation of the branches across hits, including an isolated feedback loop between the translocon and IRE1α. These studies provide insight into how the three sensors of ER homeostasis monitor distinct types of stress and highlight the ability of Perturb-seq to dissect complex cellular responses.
Keywords: CRIPSRi; CRISPR; Single-cell RNA-seq; cell-to-cell heterogeneity; genome-scale screening; single-cell genomics; unfolded protein response.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
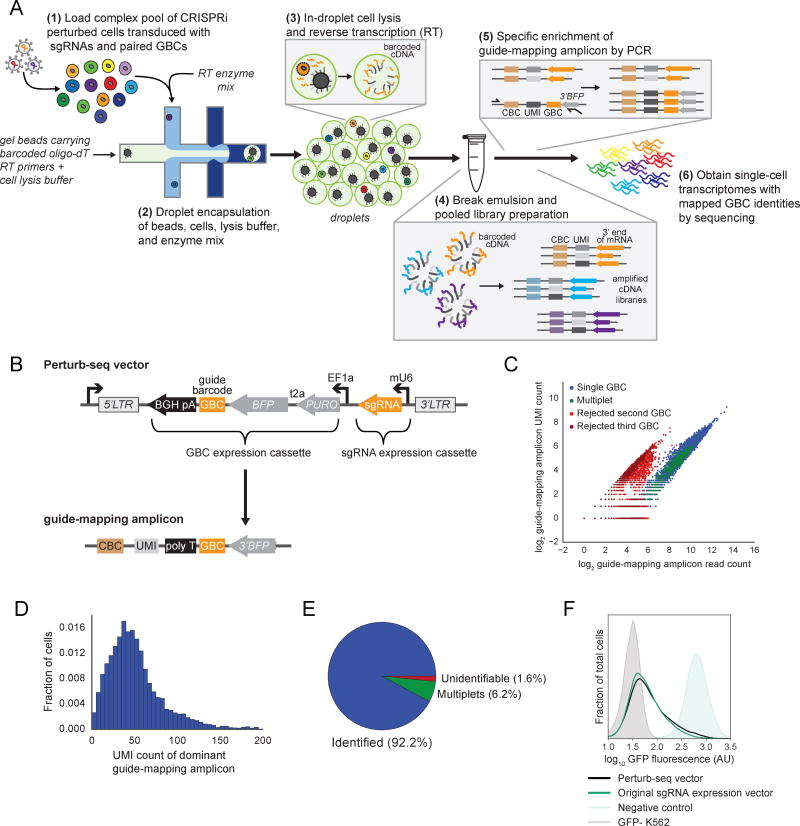
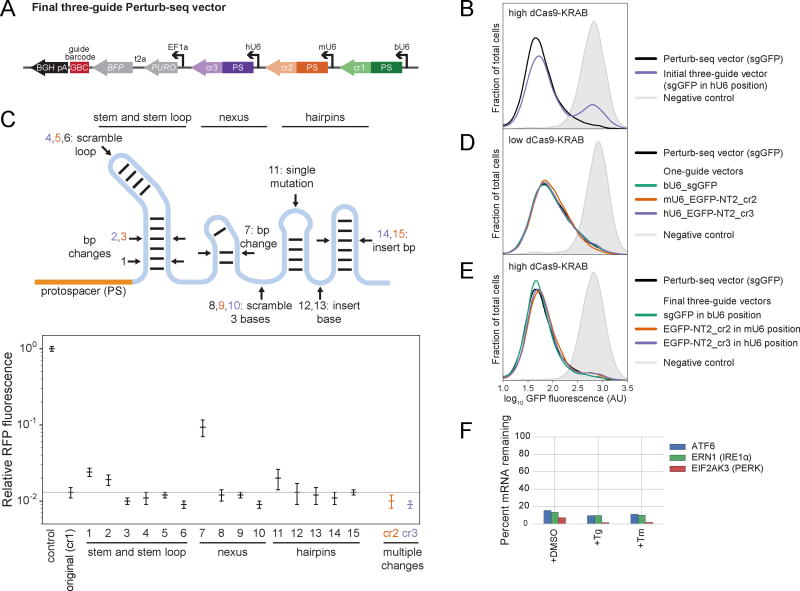
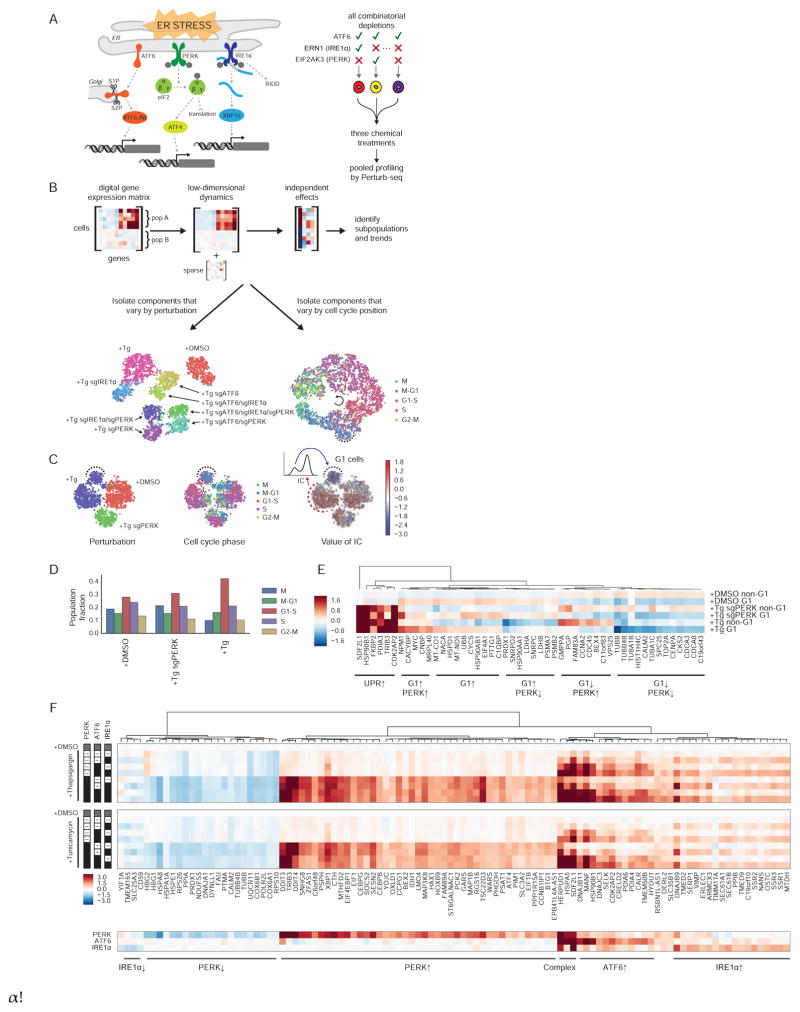
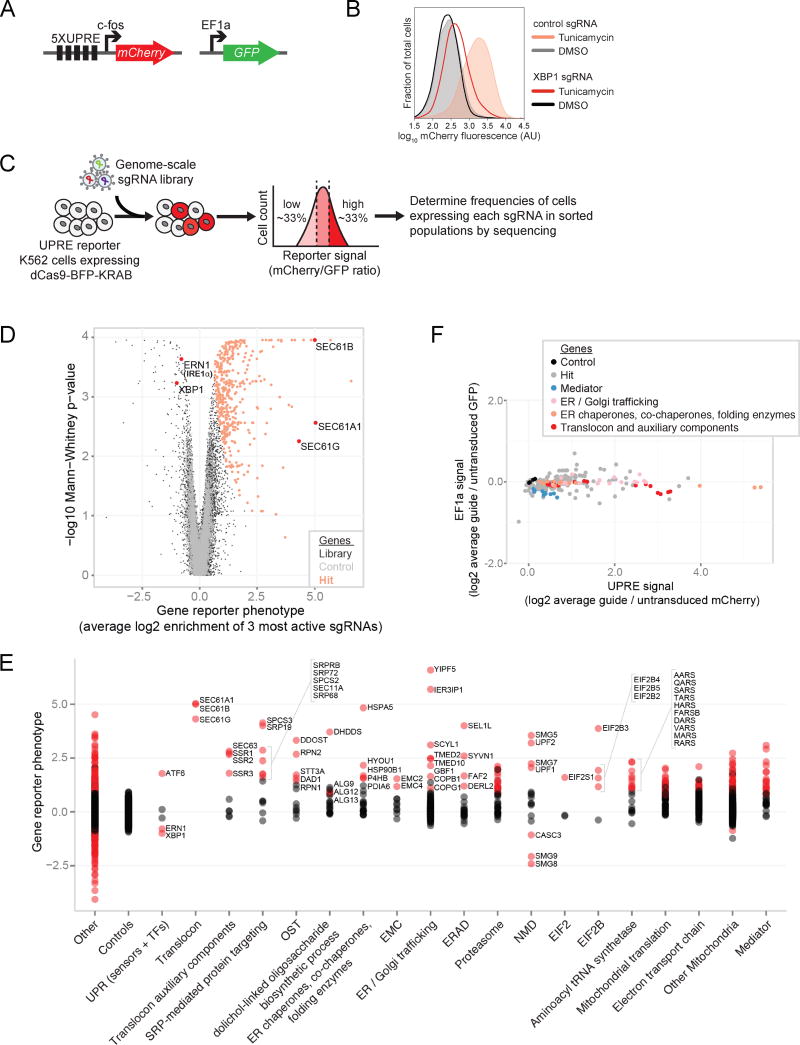
Comment in
-
Genetic screens: Combining CRISPR perturbations and RNA-seq.Nat Rev Genet. 2017 Jan 18;18(2):67. doi: 10.1038/nrg.2016.171. Nat Rev Genet. 2017. PMID: 28096529 No abstract available.
-
Genetic screening enters the single-cell era.Nat Methods. 2017 Feb 28;14(3):237-238. doi: 10.1038/nmeth.4196. Nat Methods. 2017. PMID: 28245215 No abstract available.
-
Single-minded CRISPR screening.Nat Biotechnol. 2017 Apr 11;35(4):339-340. doi: 10.1038/nbt.3849. Nat Biotechnol. 2017. PMID: 28398325 No abstract available.
References
-
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. - PubMed
-
- Candès EJ, Li X, Ma Y, Wright J. Robust principal component analysis? Journal of the ACM (JACM) 2011;58:11.
-
- Friedman J, Hastie T, Tibshirani R. Springer series in statistics. Springer; Berlin: 2001. The elements of statistical learning.
MeSH terms
Substances
Grants and funding
- U01 CA168370/CA/NCI NIH HHS/United States
- R00 CA204602/CA/NCI NIH HHS/United States
- R35 GM118061/GM/NIGMS NIH HHS/United States
- K99 CA204602/CA/NCI NIH HHS/United States
- T32 EB009383/EB/NIBIB NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 GM102790/GM/NIGMS NIH HHS/United States
- RM1 HG006193/HG/NHGRI NIH HHS/United States
- T32 GM007618/GM/NIGMS NIH HHS/United States
- F32 GM116331/GM/NIGMS NIH HHS/United States
- P50 GM102706/GM/NIGMS NIH HHS/United States
- R01 DA036858/DA/NIDA NIH HHS/United States
- R01 AG041826/AG/NIA NIH HHS/United States
- P50 HG006193/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
