

Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks
- PMID: 27984746
- PMCID: PMC5179496
- DOI: 10.1016/j.molcel.2016.10.038
Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks
Abstract
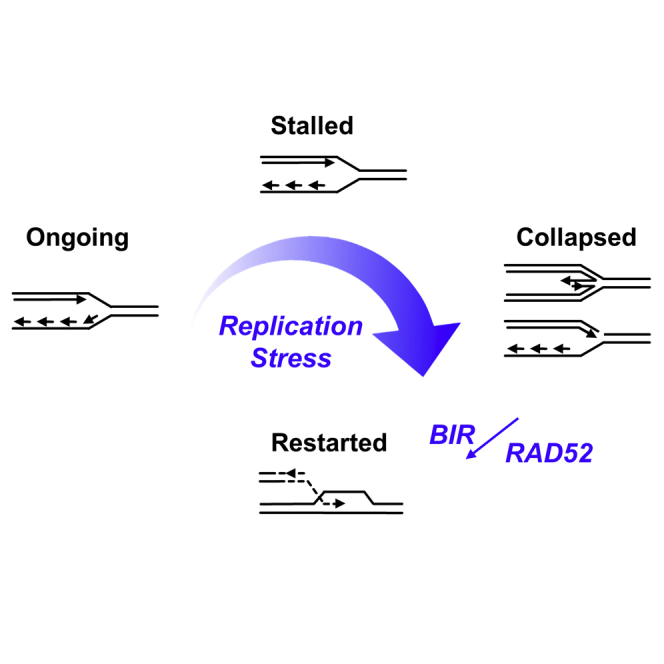
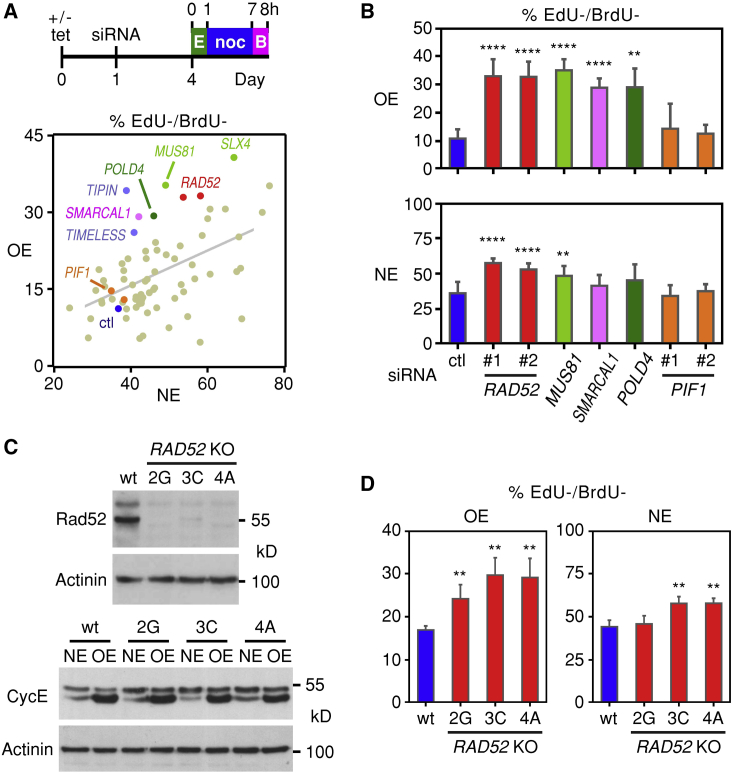
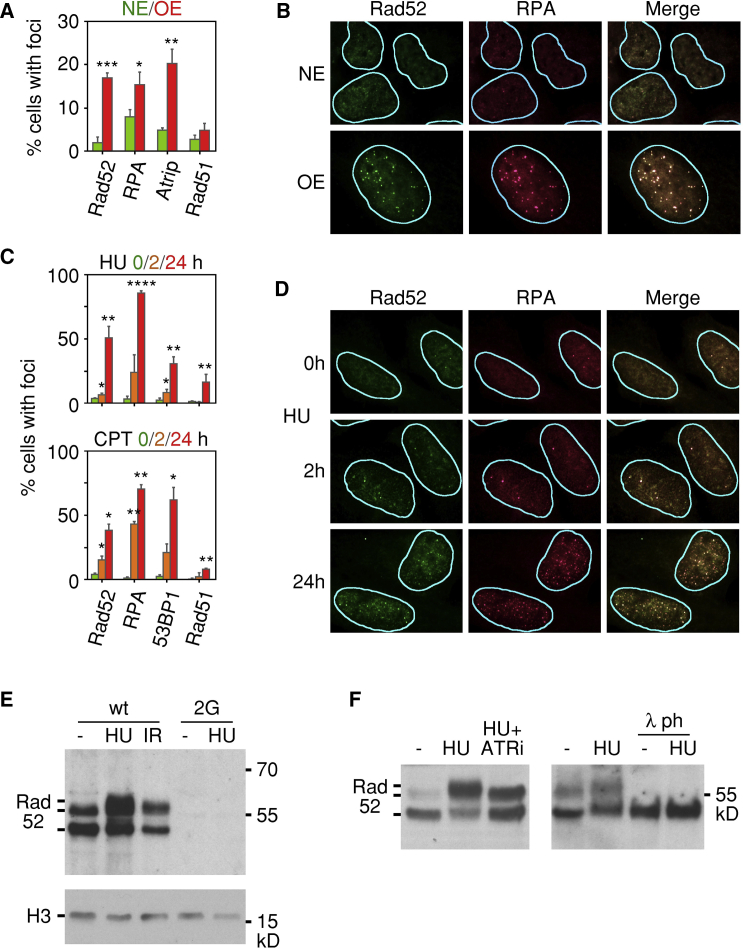
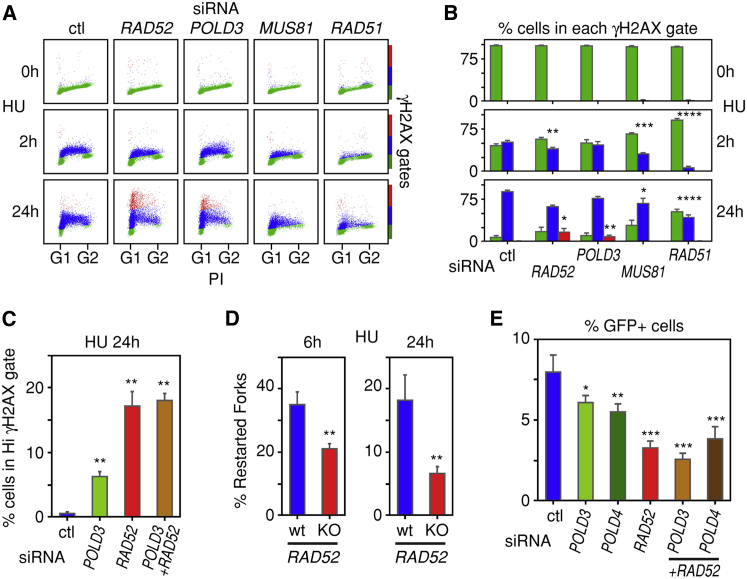
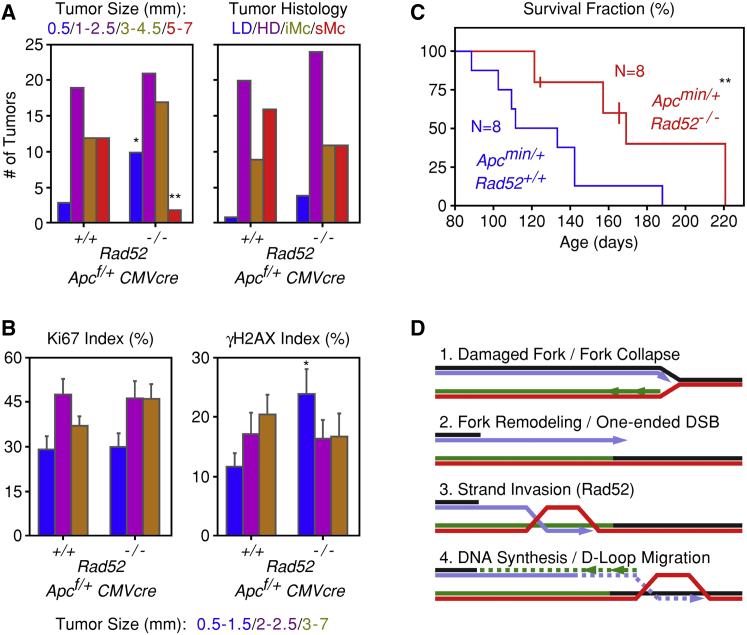
Human cancers are characterized by the presence of oncogene-induced DNA replication stress (DRS), making them dependent on repair pathways such as break-induced replication (BIR) for damaged DNA replication forks. To better understand BIR, we performed a targeted siRNA screen for genes whose depletion inhibited G1 to S phase progression when oncogenic cyclin E was overexpressed. RAD52, a gene dispensable for normal development in mice, was among the top hits. In cells in which fork collapse was induced by oncogenes or chemicals, the Rad52 protein localized to DRS foci. Depletion of Rad52 by siRNA or knockout of the gene by CRISPR/Cas9 compromised restart of collapsed forks and led to DNA damage in cells experiencing DRS. Furthermore, in cancer-prone, heterozygous APC mutant mice, homozygous deletion of the Rad52 gene suppressed tumor growth and prolonged lifespan. We therefore propose that mammalian RAD52 facilitates repair of collapsed DNA replication forks in cancer cells.
Keywords: DNA recombination; DNA replication stress; RAD52; break-induced replication; cancer.
Copyright © 2016 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Stressing Out About RAD52.Mol Cell. 2016 Dec 15;64(6):1017-1019. doi: 10.1016/j.molcel.2016.11.036. Mol Cell. 2016. PMID: 27984741
References
-
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. - PubMed
-
- Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
