

Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy
- PMID: 27989324
- PMCID: PMC5223029
- DOI: 10.1016/j.ajhg.2016.11.014
Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy
Abstract
MDH2 encodes mitochondrial malate dehydrogenase (MDH), which is essential for the conversion of malate to oxaloacetate as part of the proper functioning of the Krebs cycle. We report bi-allelic pathogenic mutations in MDH2 in three unrelated subjects presenting with early-onset generalized hypotonia, psychomotor delay, refractory epilepsy, and elevated lactate in the blood and cerebrospinal fluid. Functional studies in fibroblasts from affected subjects showed both an apparently complete loss of MDH2 levels and MDH2 enzymatic activity close to null. Metabolomics analyses demonstrated a significant concomitant accumulation of the MDH substrate, malate, and fumarate, its immediate precursor in the Krebs cycle, in affected subjects' fibroblasts. Lentiviral complementation with wild-type MDH2 cDNA restored MDH2 levels and mitochondrial MDH activity. Additionally, introduction of the three missense mutations from the affected subjects into Saccharomyces cerevisiae provided functional evidence to support their pathogenicity. Disruption of the Krebs cycle is a hallmark of cancer, and MDH2 has been recently identified as a novel pheochromocytoma and paraganglioma susceptibility gene. We show that loss-of-function mutations in MDH2 are also associated with severe neurological clinical presentations in children.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
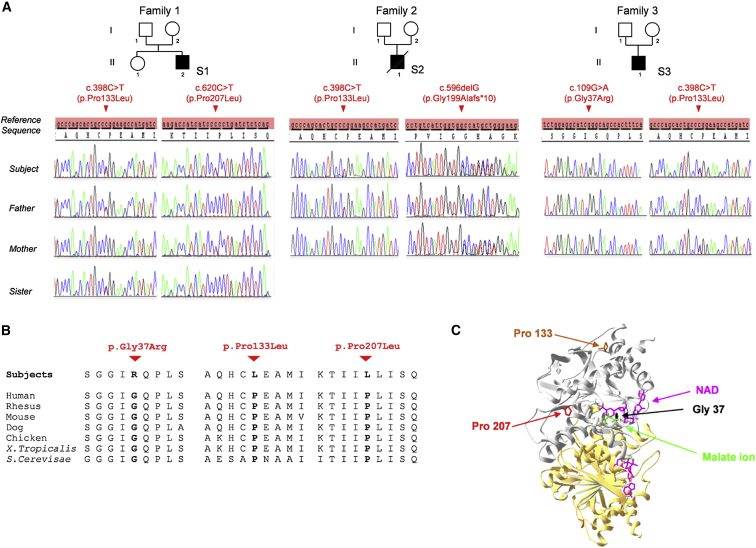
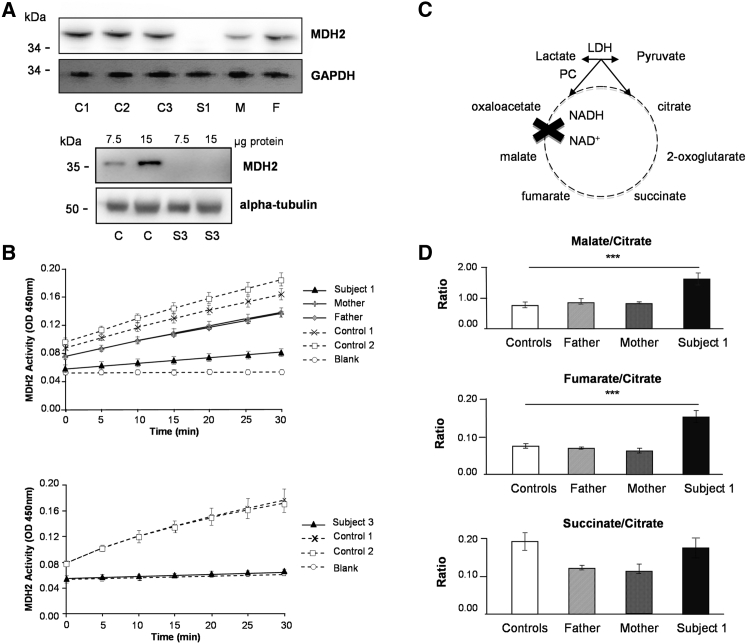
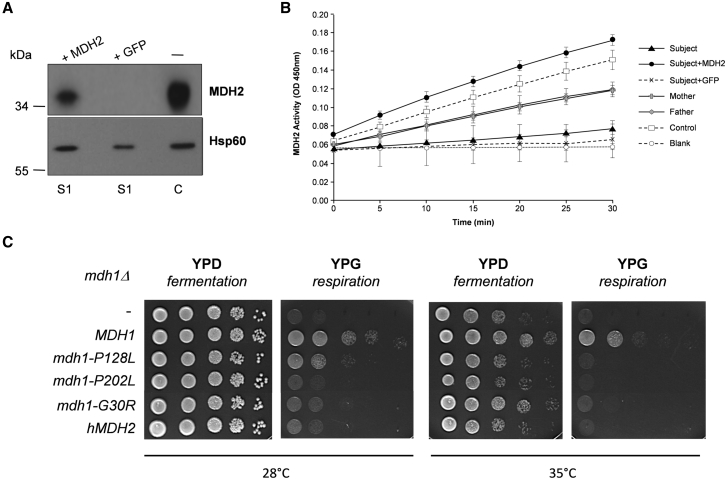
References
-
- Khurana D.S., Salganicoff L., Melvin J.J., Hobdell E.F., Valencia I., Hardison H.H., Marks H.G., Grover W.D., Legido A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics. 2008;39:8–13. - PubMed
-
- Cascón A., Comino-Méndez I., Currás-Freixes M., de Cubas A.A., Contreras L., Richter S., Peitzsch M., Mancikova V., Inglada-Pérez L., Pérez-Barrios A. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J. Natl. Cancer Inst. 2015;107 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
