

The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives
- PMID: 27990015
- PMCID: PMC5657490
- DOI: 10.1038/nrneph.2016.182
The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives
Abstract
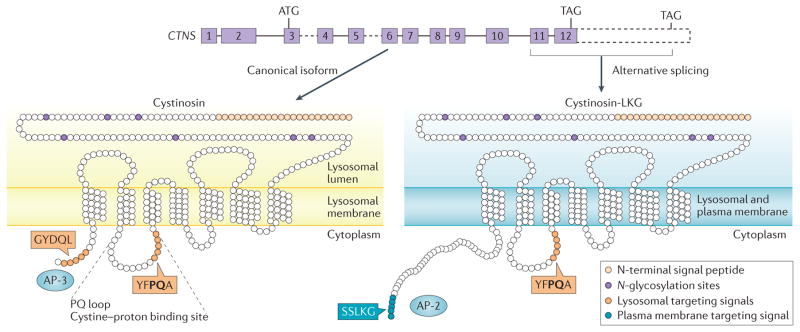
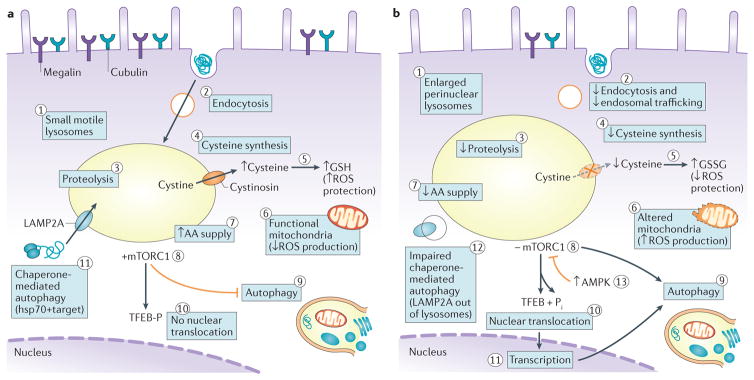
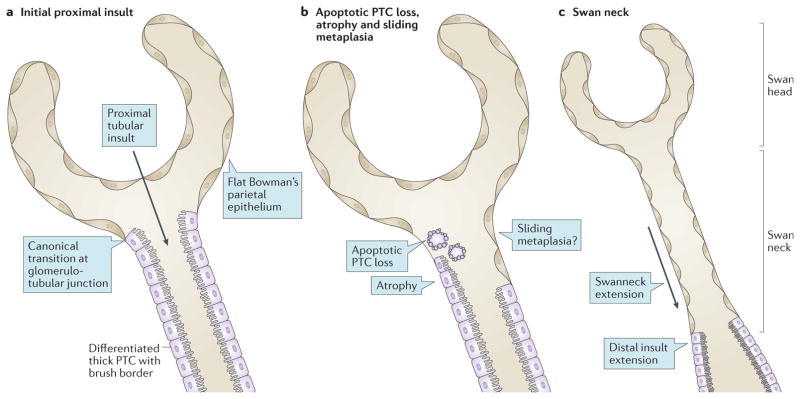
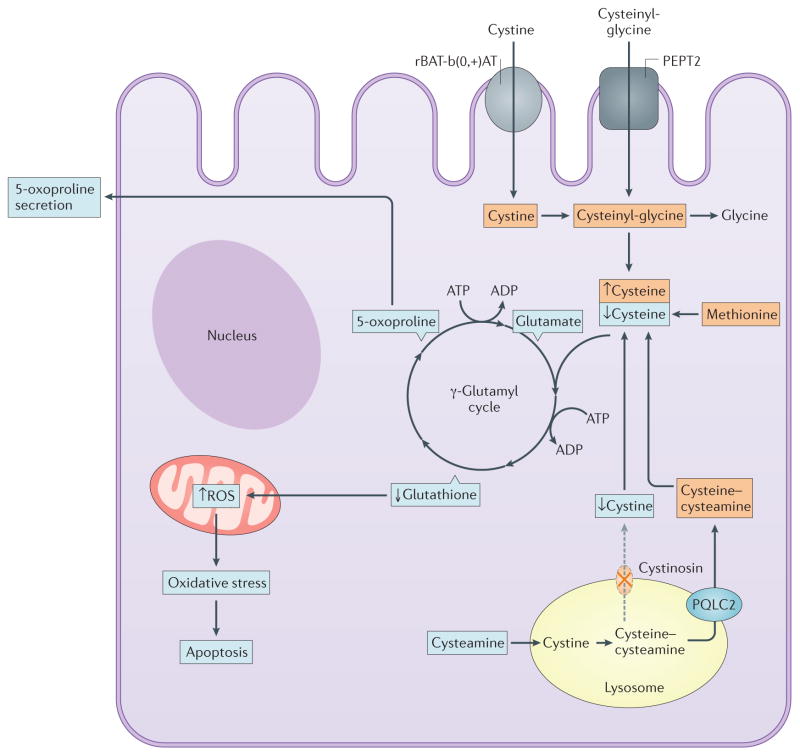
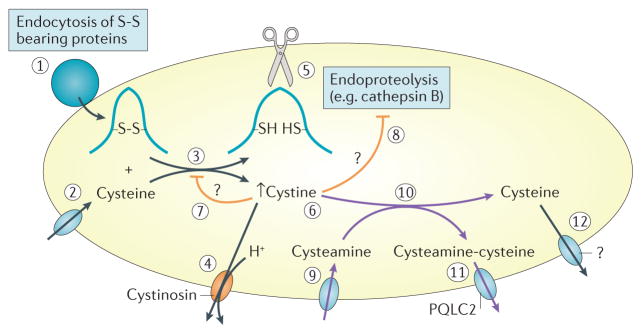
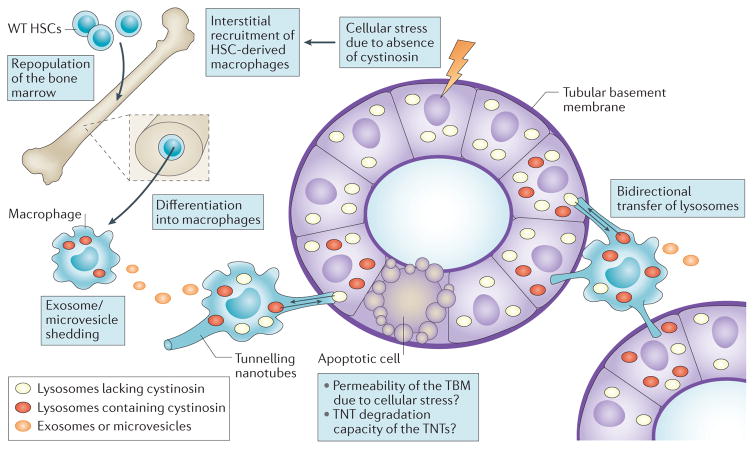
Cystinosis is an autosomal recessive metabolic disease that belongs to the family of lysosomal storage disorders. It is caused by a defect in the lysosomal cystine transporter, cystinosin, which results in an accumulation of cystine in all organs. Despite the ubiquitous expression of cystinosin, a renal Fanconi syndrome is often the first manifestation of cystinosis, usually presenting within the first year of life and characterized by the early and severe dysfunction of proximal tubule cells, highlighting the unique vulnerability of this cell type. The current therapy for cystinosis, cysteamine, facilitates lysosomal cystine clearance and greatly delays progression to kidney failure but is unable to correct the Fanconi syndrome. This Review summarizes decades of studies that have fostered a better understanding of the pathogenesis of the renal Fanconi syndrome associated with cystinosis. These studies have unraveled some of the early molecular changes that occur before the onset of tubular atrophy and identified a role for cystinosin beyond cystine transport, in endolysosomal trafficking and proteolysis, lysosomal clearance, autophagy and the regulation of energy balance. These studies have also led to the identification of new potential therapeutic targets and here, we outline the potential role of stem cell therapy for cystinosis and provide insights into the mechanism of haematopoietic stem cell-mediated kidney protection.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Fanconi G. Die nicht diabetischen glykosurien und hyperglykamiendes altem kindes. Jahrbuch Kinderheilkunde. 1931;133:257–300.
-
- De Toni G. Remarks on the relations between renal rickets (renal dwarfism) and renal diabetes. Acta Paediatr. 1933;16:479–484.
-
- Debre R, Marie J, Cleret J, Messimy R. Rachitisme tardif coexistant avec une nephrite chronique et une glycosurie. Arch Med Enfants. 1934;37:597–606.
-
- Haffner D, et al. Long-term outcome of paediatric patients with hereditary tubular disorders. Nephron. 1999;83:250–260. - PubMed
-
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–121. - PubMed
