

Toward amino acid typing for proteins in FFLUX
- PMID: 27991680
- PMCID: PMC6681421
- DOI: 10.1002/jcc.24686
Toward amino acid typing for proteins in FFLUX
Abstract
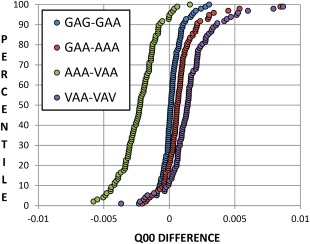
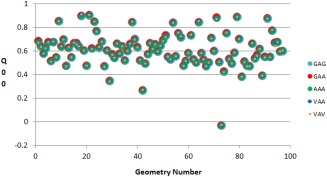
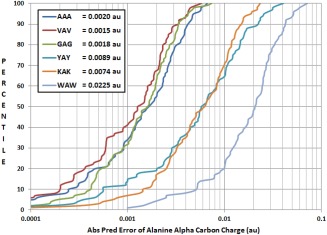
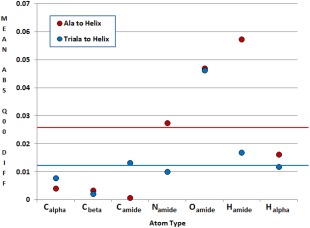
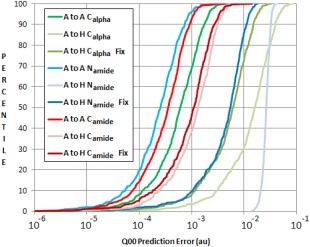
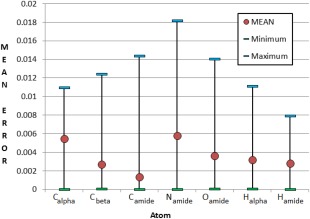
Continuing the development of the FFLUX, a multipolar polarizable force field driven by machine learning, we present a modern approach to atom-typing and building transferable models for predicting atomic properties in proteins. Amino acid atomic charges in a peptide chain respond to the substitution of a neighboring residue and this response can be categorized in a manner similar to atom-typing. Using a machine learning method called kriging, we are able to build predictive models for an atom that is defined, not only by its local environment, but also by its neighboring residues, for a minimal additional computational cost. We found that prediction errors were up to 11 times lower when using a model specific to the correct group of neighboring residues, with a mean prediction of ∼0.0015 au. This finding suggests that atoms in a force field should be defined by more than just their immediate atomic neighbors. When comparing an atom in a single alanine to an analogous atom in a deca-alanine helix, the mean difference in charge is 0.026 au. Meanwhile, the same difference between a trialanine and a deca-alanine helix is only 0.012 au. When compared to deca-alanine models, the transferable models are up to 20 times faster to train, and require significantly less ab initio calculation, providing a practical route to modeling large biological systems. © 2016 The Authors. Journal of Computational Chemistry Published by Wiley Periodicals, Inc.
Keywords: QTAIM; atomic charge; force field design; kriging; machine learning; peptides; quantum chemical topology.
© 2016 The Authors. Journal of Computational Chemistry Published by Wiley Periodicals, Inc.
Figures
References
-
- Wishart D. S., Bigam C. G., Holm A., Hodges R. S., Sykes B. D., J. Biomol. NMR 1994, 5, 67. - PubMed
-
- Monaco G., Int. J. Quantum Chem. 1998, 68, 201.
-
- Thomas A., Milon A., Brasseur R., Proteins Struct. Funct. Bioinform. 2004, 56, 102. - PubMed
-
- Rypniewski W. R., Perrakis A., Vorgias C. E., Wilson K. S., Protein Eng. 1994, 7, 57. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
