

Extreme mutation bias and high AT content in Plasmodium falciparum
- PMID: 27994033
- PMCID: PMC5389722
- DOI: 10.1093/nar/gkw1259
Extreme mutation bias and high AT content in Plasmodium falciparum
Abstract
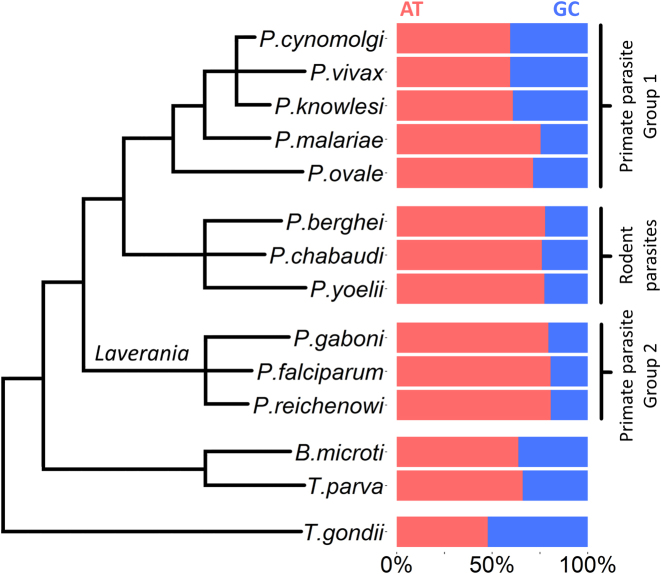
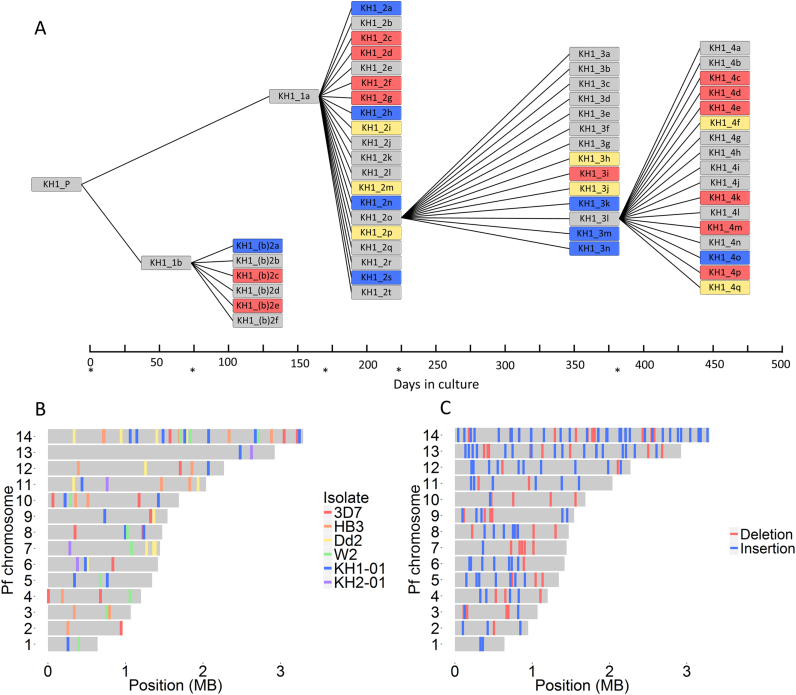
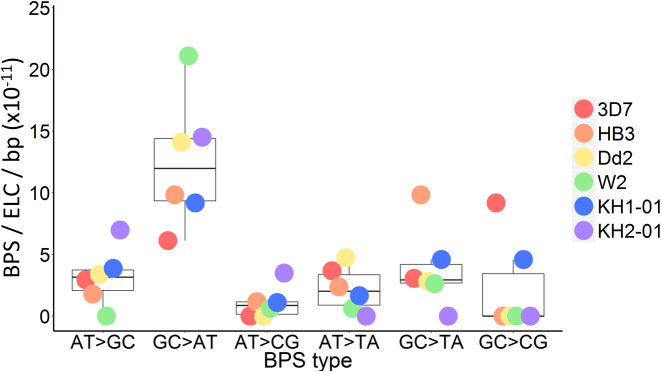
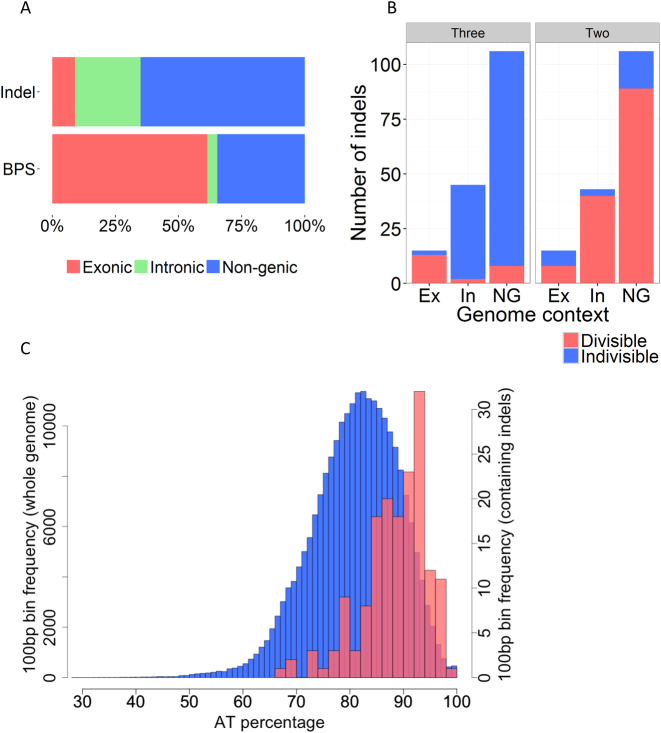
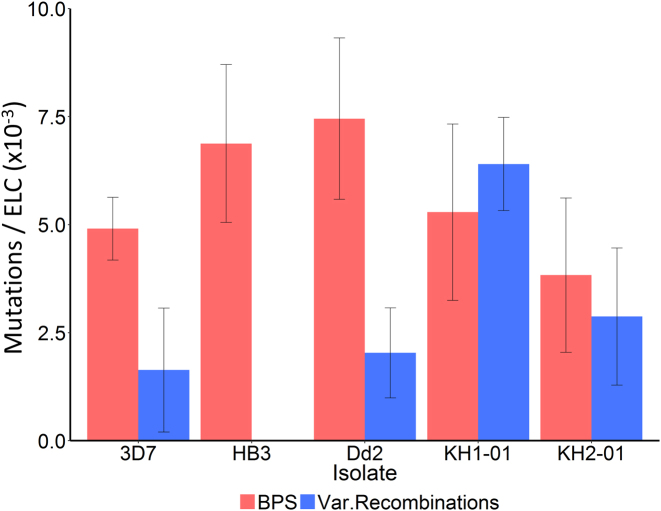
For reasons that remain unknown, the Plasmodium falciparum genome has an exceptionally high AT content compared to other Plasmodium species and eukaryotes in general - nearly 80% in coding regions and approaching 90% in non-coding regions. Here, we examine how this phenomenon relates to genome-wide patterns of de novo mutation. Mutation accumulation experiments were performed by sequential cloning of six P. falciparum isolates growing in human erythrocytes in vitro for 4 years, with 279 clones sampled for whole genome sequencing at different time points. Genome sequence analysis of these samples revealed a significant excess of G:C to A:T transitions compared to other types of nucleotide substitution, which would naturally cause AT content to equilibrate close to the level seen across the P. falciparum reference genome (80.6% AT). These data also uncover an extremely high rate of small indel mutation relative to other species, primarily associated with repetitive AT-rich sequences, in addition to larger-scale structural rearrangements focused in antigen-coding var genes. In conclusion, high AT content in P. falciparum is driven by a systematic mutational bias and ultimately leads to an unusual level of microstructural plasticity, raising the question of whether this contributes to adaptive evolution.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Nikbakht H., Xia X., Hickey D.A.. The evolution of genomic GC content undergoes a rapid reversal within the genus Plasmodium. Genome. 2015; 57:507–511. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
