

Focal Adhesion Kinase-mediated Phosphorylation of Beclin1 Protein Suppresses Cardiomyocyte Autophagy and Initiates Hypertrophic Growth
- PMID: 27994061
- PMCID: PMC5313082
- DOI: 10.1074/jbc.M116.758268
Focal Adhesion Kinase-mediated Phosphorylation of Beclin1 Protein Suppresses Cardiomyocyte Autophagy and Initiates Hypertrophic Growth
Abstract
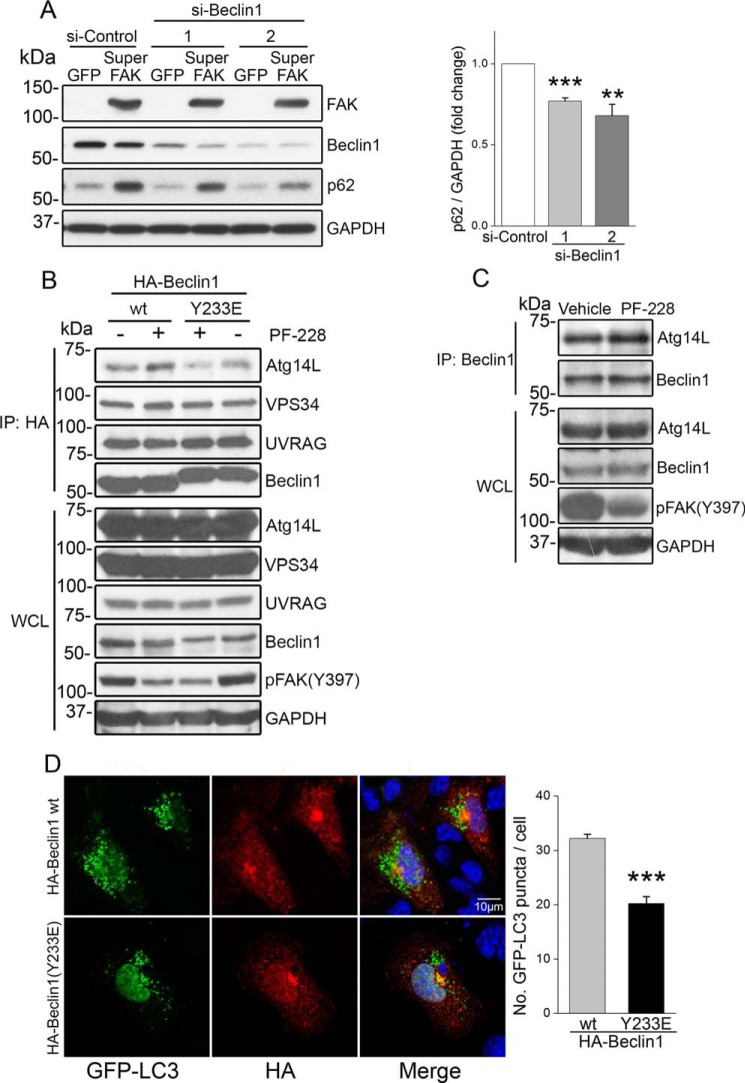
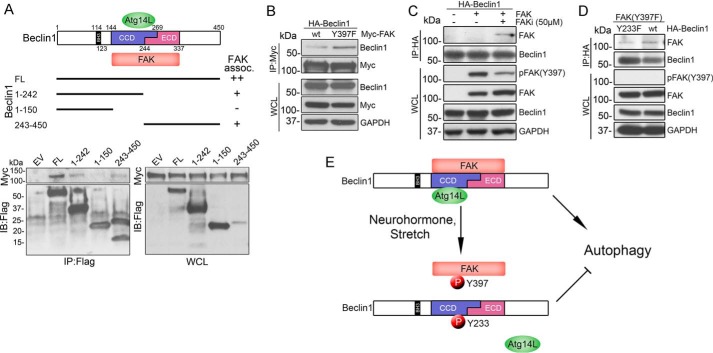
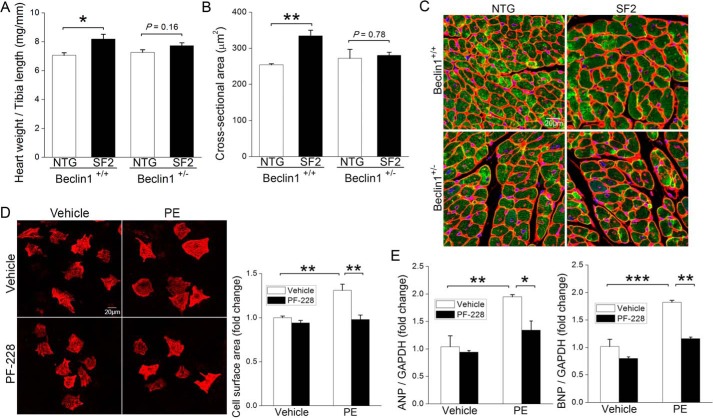
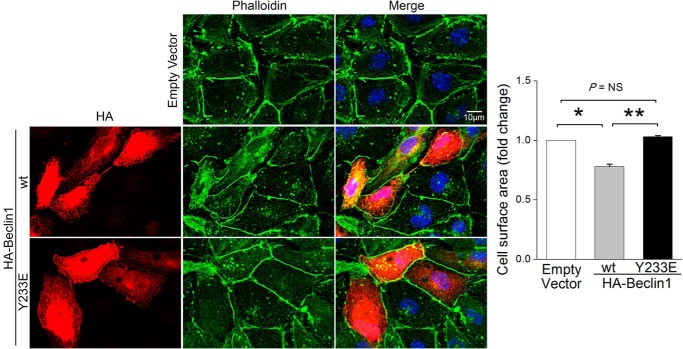
Autophagy is an evolutionarily conserved intracellular degradation/recycling system that is essential for cellular homeostasis but is dysregulated in a number of diseases, including myocardial hypertrophy. Although it is clear that limiting or accelerating autophagic flux can result in pathological cardiac remodeling, the physiological signaling pathways that fine-tune cardiac autophagy are poorly understood. Herein, we demonstrated that stimulation of cardiomyocytes with phenylephrine (PE), a well known hypertrophic agonist, suppresses autophagy and that activation of focal adhesion kinase (FAK) is necessary for PE-stimulated autophagy suppression and subsequent initiation of hypertrophic growth. Mechanistically, we showed that FAK phosphorylates Beclin1, a core autophagy protein, on Tyr-233 and that this post-translational modification limits Beclin1 association with Atg14L and reduces Beclin1-dependent autophagosome formation. Remarkably, although ectopic expression of wild-type Beclin1 promoted cardiomyocyte atrophy, expression of a Y233E phosphomimetic variant of Beclin1 failed to affect cardiomyocyte size. Moreover, genetic depletion of Beclin1 attenuated PE-mediated/FAK-dependent initiation of myocyte hypertrophy in vivo Collectively, these findings identify FAK as a novel negative regulator of Beclin1-mediated autophagy and indicate that this pathway can facilitate the promotion of compensatory hypertrophic growth. This novel mechanism to limit Beclin1 activity has important implications for treating a variety of pathologies associated with altered autophagic flux.
Keywords: Beclin-1 (BECN1); PTK2 protein tyrosine kinase 2 (PTK2) (focal adhesion kinase) (FAK); atrophy; autophagy; cardiac hypertrophy; muscle atrophy.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
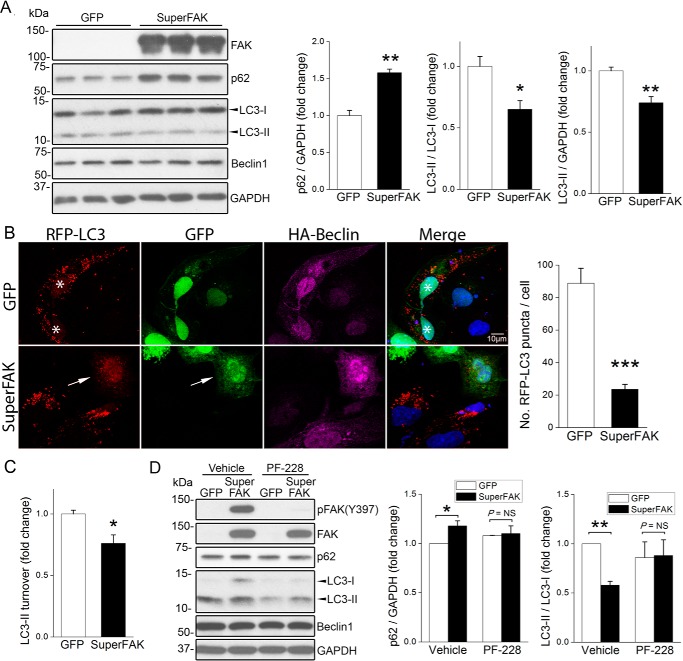
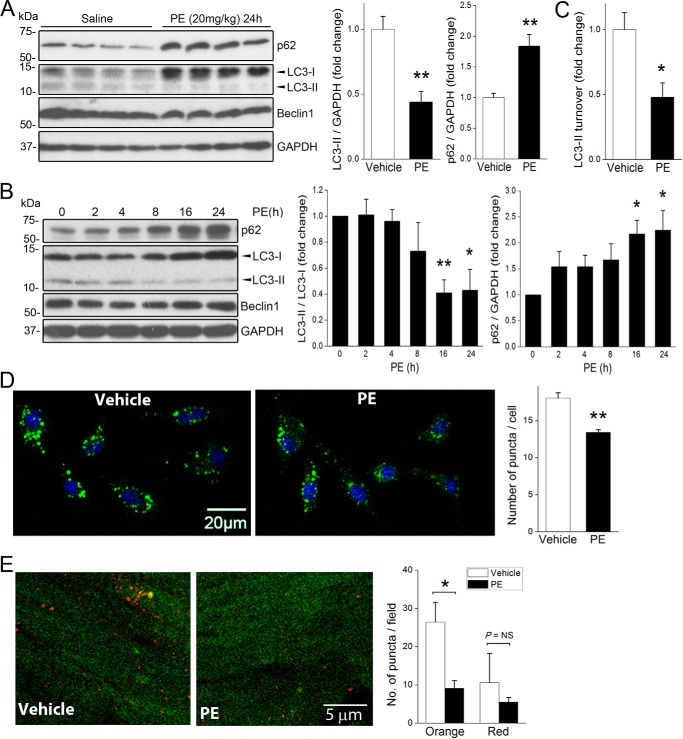
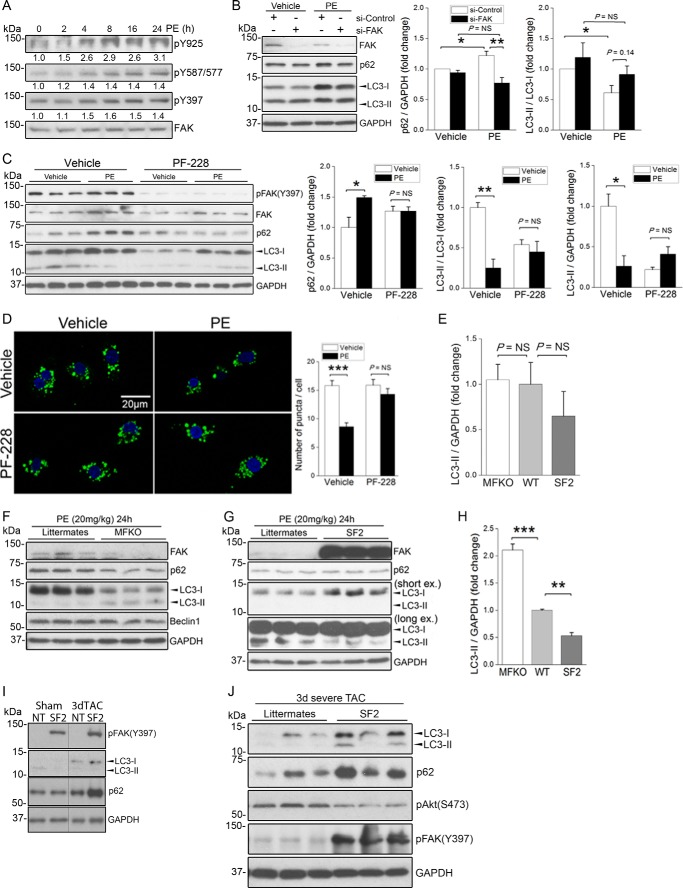
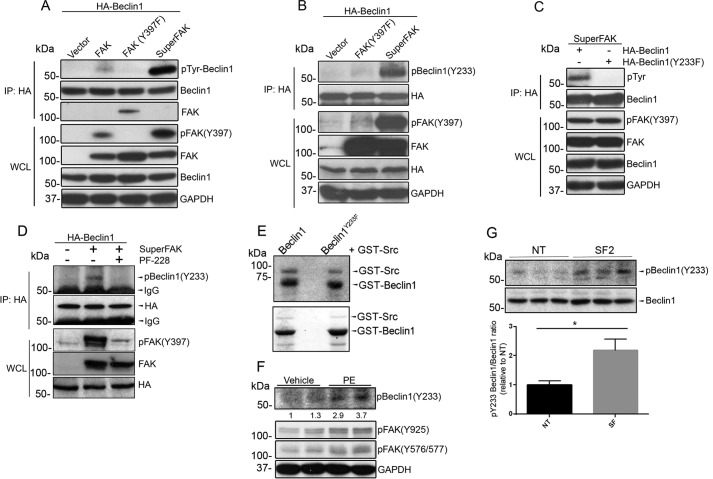
References
-
- Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., and Otsu K. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 - PubMed
-
- Kanamori H., Takemura G., Goto K., Maruyama R., Tsujimoto A., Ogino A., Takeyama T., Kawaguchi T., Watanabe T., Fujiwara T., Fujiwara H., Seishima M., and Minatoguchi S. (2011) The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 91, 330–339 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
