

Emerging Roles for the Immune System in Traumatic Brain Injury
- PMID: 27994591
- PMCID: PMC5137185
- DOI: 10.3389/fimmu.2016.00556
Emerging Roles for the Immune System in Traumatic Brain Injury
Abstract
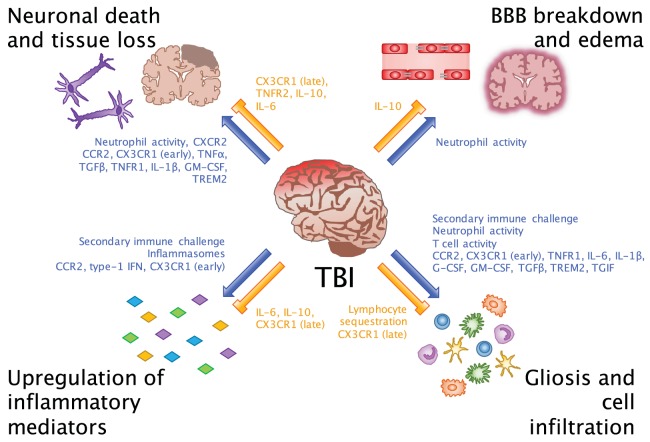
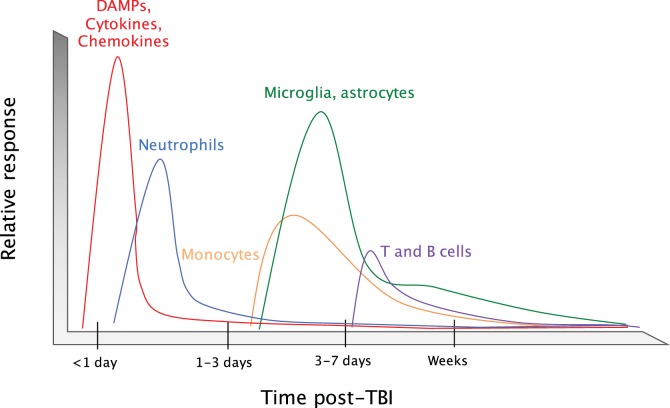
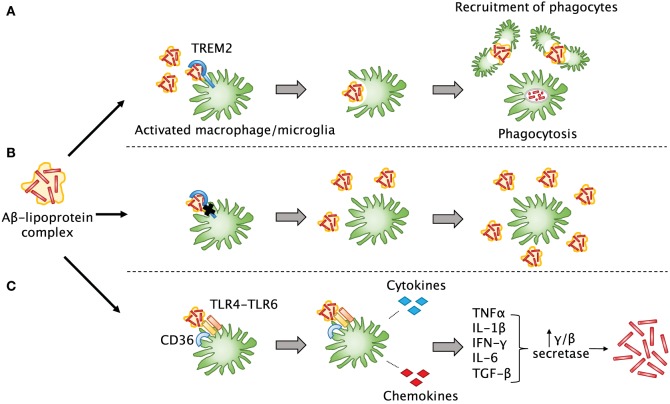
Traumatic brain injury (TBI) affects an ever-growing population of all ages with long-term consequences on health and cognition. Many of the issues that TBI patients face are thought to be mediated by the immune system. Primary brain damage that occurs at the time of injury can be exacerbated and prolonged for months or even years by chronic inflammatory processes, which can ultimately lead to secondary cell death, neurodegeneration, and long-lasting neurological impairment. Researchers have turned to rodent models of TBI in order to understand how inflammatory cells and immunological signaling regulate the post-injury response and recovery mechanisms. In addition, the development of numerous methods to manipulate genes involved in inflammation has recently expanded the possibilities of investigating the immune response in TBI models. As results from these studies accumulate, scientists have started to link cells and signaling pathways to pro- and anti-inflammatory processes that may contribute beneficial or detrimental effects to the injured brain. Moreover, emerging data suggest that targeting aspects of the immune response may offer promising strategies to treat TBI. This review will cover insights gained from studies that approach TBI research from an immunological perspective and will summarize our current understanding of the involvement of specific immune cell types and cytokines in TBI pathogenesis.
Keywords: cytokine; inflammasome; innate immunology; microglia; neurodegeneration; neuroinflammation; neuroprotection; traumatic brain injury.
Figures
References
-
- Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, et al. Surveillance for traumatic brain injury-related deaths – United States, 1997-2007. MMWR Surveill Summ (2011) 60(5):1–32. - PubMed
-
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol (1991) 20(Suppl 2):S28–35.10.1093/ije/20.Supplement_2.S28 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials