

IL-7 Receptor Mutations and Steroid Resistance in Pediatric T cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study
- PMID: 27997540
- PMCID: PMC5172551
- DOI: 10.1371/journal.pmed.1002200
IL-7 Receptor Mutations and Steroid Resistance in Pediatric T cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study
Abstract
Background: Pediatric acute lymphoblastic leukemia (ALL) is the most common childhood cancer and the leading cause of cancer-related mortality in children. T cell ALL (T-ALL) represents about 15% of pediatric ALL cases and is considered a high-risk disease. T-ALL is often associated with resistance to treatment, including steroids, which are currently the cornerstone for treating ALL; moreover, initial steroid response strongly predicts survival and cure. However, the cellular mechanisms underlying steroid resistance in T-ALL patients are poorly understood. In this study, we combined various genomic datasets in order to identify candidate genetic mechanisms underlying steroid resistance in children undergoing T-ALL treatment.
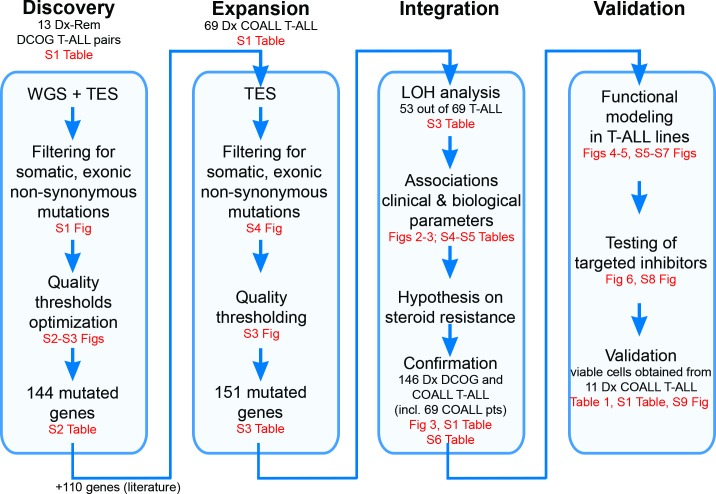
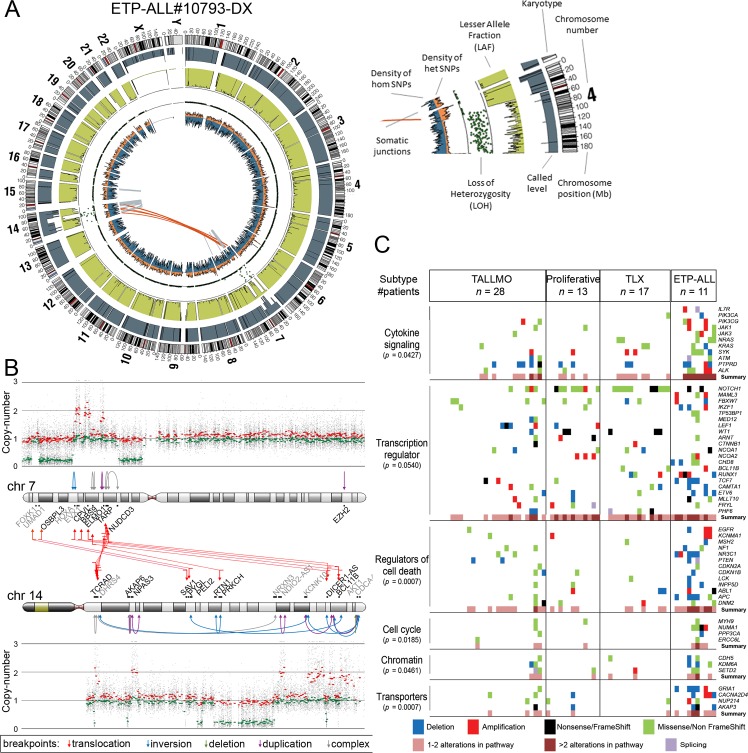
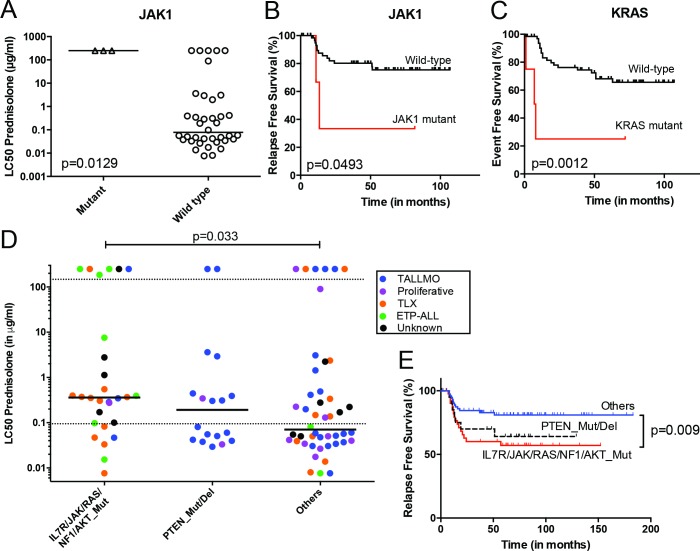
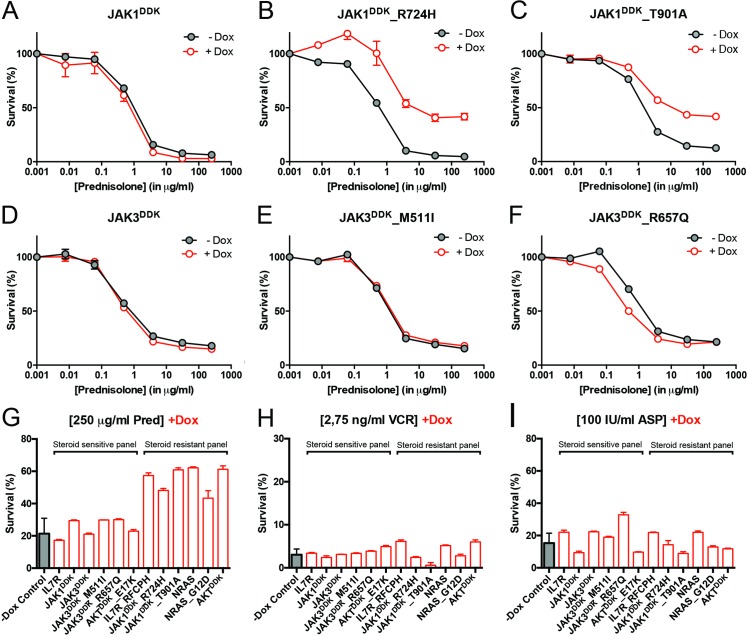
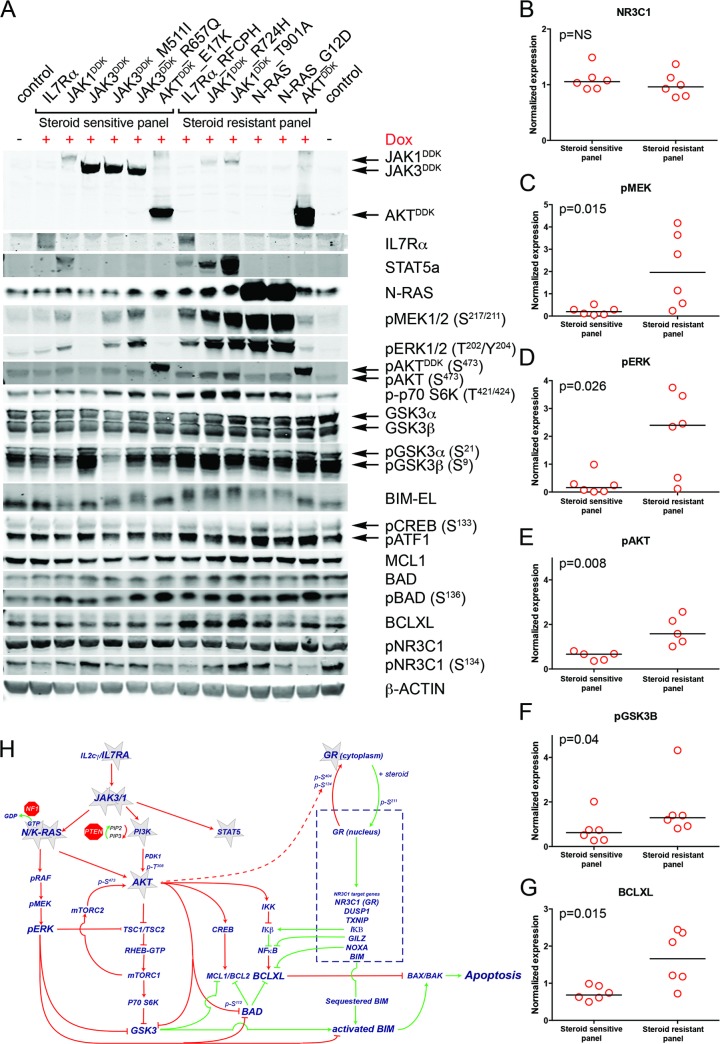
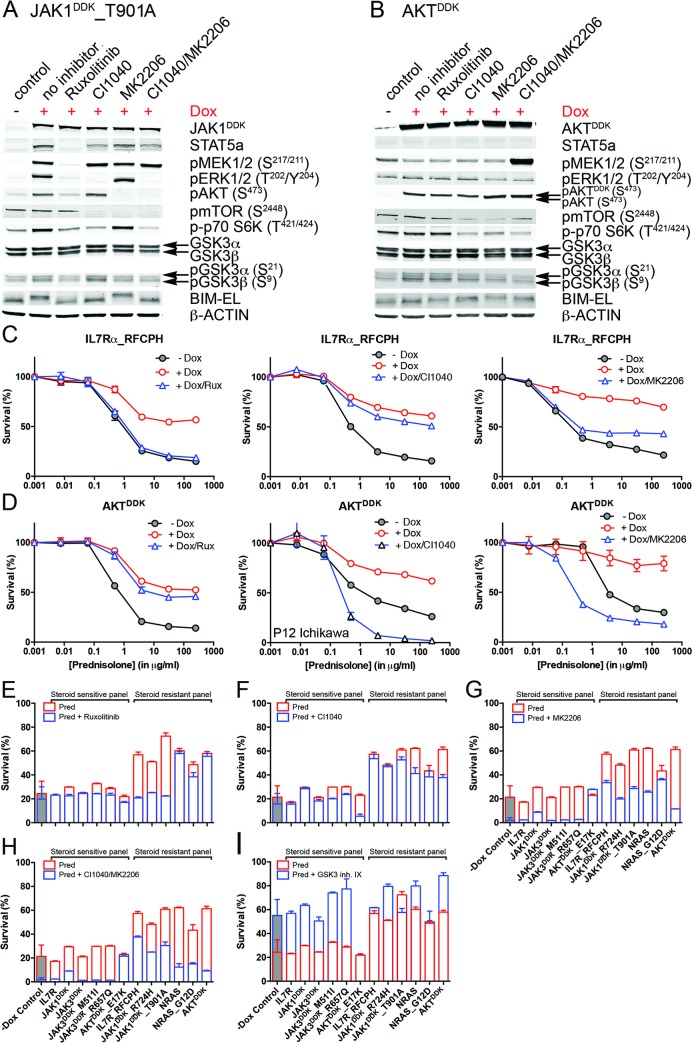
Methods and findings: We performed whole genome sequencing on paired pre-treatment (diagnostic) and post-treatment (remission) samples from 13 patients, and targeted exome sequencing of pre-treatment samples from 69 additional T-ALL patients. We then integrated mutation data with copy number data for 151 mutated genes, and this integrated dataset was tested for associations of mutations with clinical outcomes and in vitro drug response. Our analysis revealed that mutations in JAK1 and KRAS, two genes encoding components of the interleukin 7 receptor (IL7R) signaling pathway, were associated with steroid resistance and poor outcome. We then sequenced JAK1, KRAS, and other genes in this pathway, including IL7R, JAK3, NF1, NRAS, and AKT, in these 69 T-ALL patients and a further 77 T-ALL patients. We identified mutations in 32% (47/146) of patients, the majority of whom had a specific T-ALL subtype (early thymic progenitor ALL or TLX). Based on the outcomes of these patients and their prednisolone responsiveness measured in vitro, we then confirmed that these mutations were associated with both steroid resistance and poor outcome. To explore how these mutations in IL7R signaling pathway genes cause steroid resistance and subsequent poor outcome, we expressed wild-type and mutant IL7R signaling molecules in two steroid-sensitive T-ALL cell lines (SUPT1 and P12 Ichikawa cells) using inducible lentiviral expression constructs. We found that expressing mutant IL7R, JAK1, or NRAS, or wild-type NRAS or AKT, specifically induced steroid resistance without affecting sensitivity to vincristine or L-asparaginase. In contrast, wild-type IL7R, JAK1, and JAK3, as well as mutant JAK3 and mutant AKT, had no effect. We then performed a functional study to examine the mechanisms underlying steroid resistance and found that, rather than changing the steroid receptor's ability to activate downstream targets, steroid resistance was associated with strong activation of MEK-ERK and AKT, downstream components of the IL7R signaling pathway, thereby inducing a robust antiapoptotic response by upregulating MCL1 and BCLXL expression. Both the MEK-ERK and AKT pathways also inactivate BIM, an essential molecule for steroid-induced cell death, and inhibit GSK3B, an important regulator of proapoptotic BIM. Importantly, treating our cell lines with IL7R signaling inhibitors restored steroid sensitivity. To address clinical relevance, we treated primary T-ALL cells obtained from 11 patients with steroids either alone or in combination with IL7R signaling inhibitors; we found that including a MEK, AKT, mTOR, or dual PI3K/mTOR inhibitor strongly increased steroid-induced cell death. Therefore, combining these inhibitors with steroid treatment may enhance steroid sensitivity in patients with ALL. The main limitation of our study was the modest cohort size, owing to the very low incidence of T-ALL.
Conclusions: Using an unbiased sequencing approach, we found that specific mutations in IL7R signaling molecules underlie steroid resistance in T-ALL. Future prospective clinical studies should test the ability of inhibitors of MEK, AKT, mTOR, or PI3K/mTOR to restore or enhance steroid sensitivity and improve clinical outcome.
Conflict of interest statement
RCB and GJRZ are founders and shareholders of Netherlands Translational Research Center B.V. The other authors have declared that no competing interests exist.
Figures
References
-
- Pieters R, Huismans DR, Loonen AH, Hahlen K, van der Does-van den Berg A, van Wering ER, et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet. 1991;338(8764):399–403. - PubMed
-
- Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65(21):9712–8. 10.1158/0008-5472.CAN-05-1227 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
