

The Genetic Architecture of Major Depressive Disorder in Han Chinese Women
- PMID: 28002544
- PMCID: PMC5319866
- DOI: 10.1001/jamapsychiatry.2016.3578
The Genetic Architecture of Major Depressive Disorder in Han Chinese Women
Abstract
Importance: Despite the moderate, well-demonstrated heritability of major depressive disorder (MDD), there has been limited success in identifying replicable genetic risk loci, suggesting a complex genetic architecture. Research is needed to quantify the relative contribution of classes of genetic variation across the genome to inform future genetic studies of MDD.
Objectives: To apply aggregate genetic risk methods to clarify the genetic architecture of MDD by estimating and partitioning heritability by chromosome, minor allele frequency, and functional annotations and to test for enrichment of rare deleterious variants.
Design, setting, and participants: The CONVERGE (China, Oxford, and Virginia Commonwealth University Experimental Research on Genetic Epidemiology) study collected data on 5278 patients with recurrent MDD from 58 provincial mental health centers and psychiatric departments of general medical hospitals in 45 cities and 23 provinces of China. Screened controls (n = 5196) were recruited from a range of locations, including general hospitals and local community centers. Data were collected from August 1, 2008, to October 31, 2012.
Main outcomes and measures: Genetic risk for liability to recurrent MDD was partitioned using sparse whole-genome sequencing.
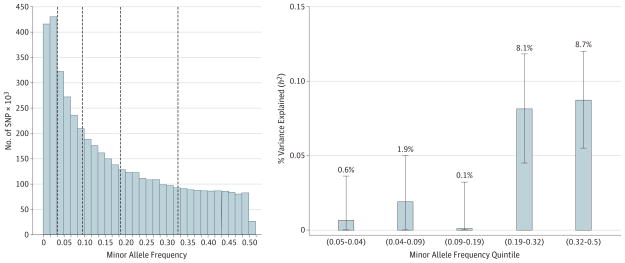
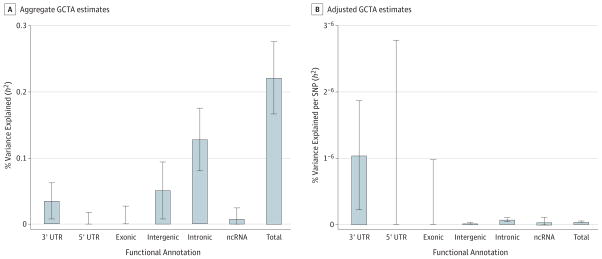
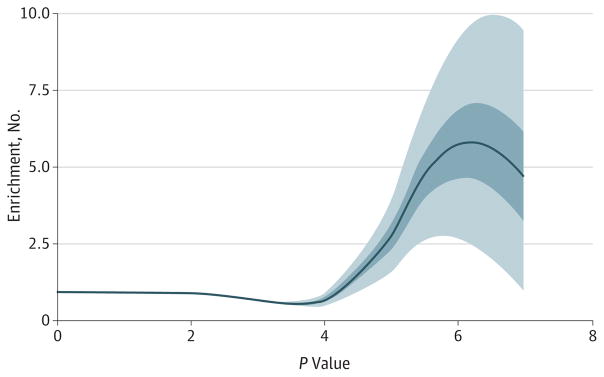
Results: In aggregate, common single-nucleotide polymorphisms (SNPs) explained between 20% and 29% of the variance in MDD risk, and the heritability in MDD explained by each chromosome was proportional to its length (r = 0.680; P = .0003), supporting a common polygenic etiology. Partitioning heritability by minor allele frequency indicated that the variance explained was distributed across the allelic frequency spectrum, although relatively common SNPs accounted for a disproportionate fraction of risk. Partitioning by genic annotation indicated a greater contribution of SNPs in protein-coding regions and within 3'-UTR regions of genes. Enrichment of SNPs associated with DNase I-hypersensitive sites was also found in many tissue types, including brain tissue. Examining burden scores from singleton exonic SNPs predicted to be deleterious indicated that cases had significantly more mutations than controls (odds ratio, 1.009; 95% CI, 1.003-1.014; P = .003), including those occurring in genes expressed in the brain (odds ratio, 1.011; 95% CI, 1.003-1.018; P = .004) and within nuclear-encoded genes with mitochondrial gene products (odds ratio, 1.075; 95% CI, 1.018-1.135; P = .009).
Conclusions and relevance: Results support a complex etiology for MDD and highlight the value of analyzing components of heritability to clarify genetic architecture.
Conflict of interest statement
Figures
References
-
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–1562. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
