

Concomitant inhibition of receptor tyrosine kinases and downstream AKT synergistically inhibited growth of KRAS/BRAF mutant colorectal cancer cells
- PMID: 28002807
- PMCID: PMC5354887
- DOI: 10.18632/oncotarget.14009
Concomitant inhibition of receptor tyrosine kinases and downstream AKT synergistically inhibited growth of KRAS/BRAF mutant colorectal cancer cells
Abstract
Receptor tyrosine kinase (RTK) signaling pathways are frequently activated in cancer cells due to mutations of RTKs and/or their downstream signaling proteins such as KRAS and BRAF. About 40% colorectal cancers (CRCs) contain KRAS or BRAF mutant genes and are resistant to treatments with individual inhibitors of RTKs, AKT, MEK, or BRAF. Therefore, an understanding of the molecular mechanisms of the drug resistance is necessary for developing effective strategies to treat the diseases. Here we report the discovery of an AKT/ERK reactivation mechanism that account for the cancer cell resistance to the AKT and MEK inhibitors treatments. The reactivations of AKT and ERK after the AKT or MEK inhibitor treatment were caused by a relief of an AKT or ERK-mediated feedback inhibition of the RTKs and/or their downstream pathways. A combination of RTK inhibitors, based on the RTK activation/phosphorylation profile, synergized with the AKT inhibitor, but not the MEK inhibitor, to completely inhibit the AKT phosphorylation and to block the growth of KRAS/BRAF mutant CRC cells. These results underscored the importance of AKT and the AKT feedback signaling to cancer cell growth and offered a novel therapeutic approach for the treatment of KRAS/BRAF mutant CRC cells.
Keywords: AKT; RAS/RAF; colorectal cancer; drug combination; receptor tyrosine kinases.
Conflict of interest statement
The authors disclose no potential conflicts of interest.
Figures
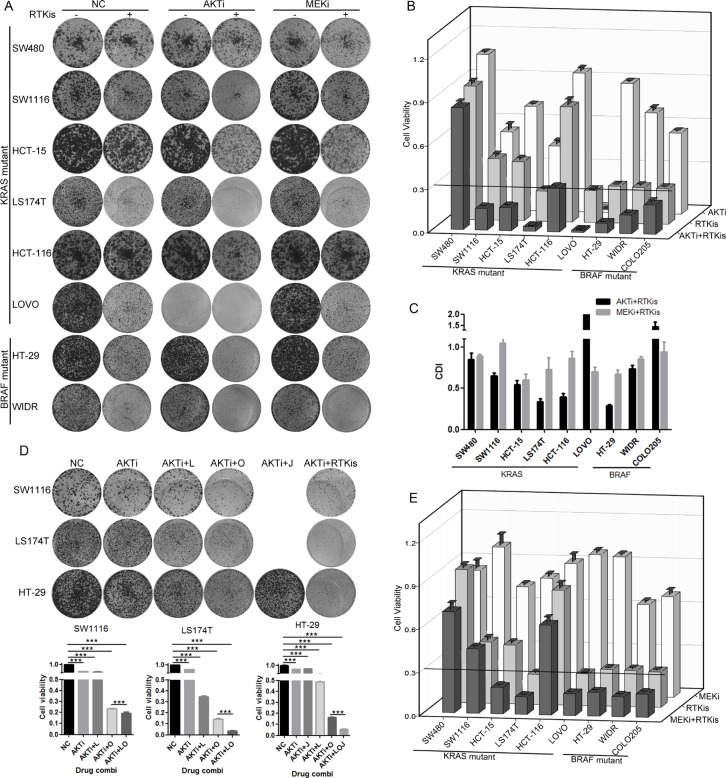
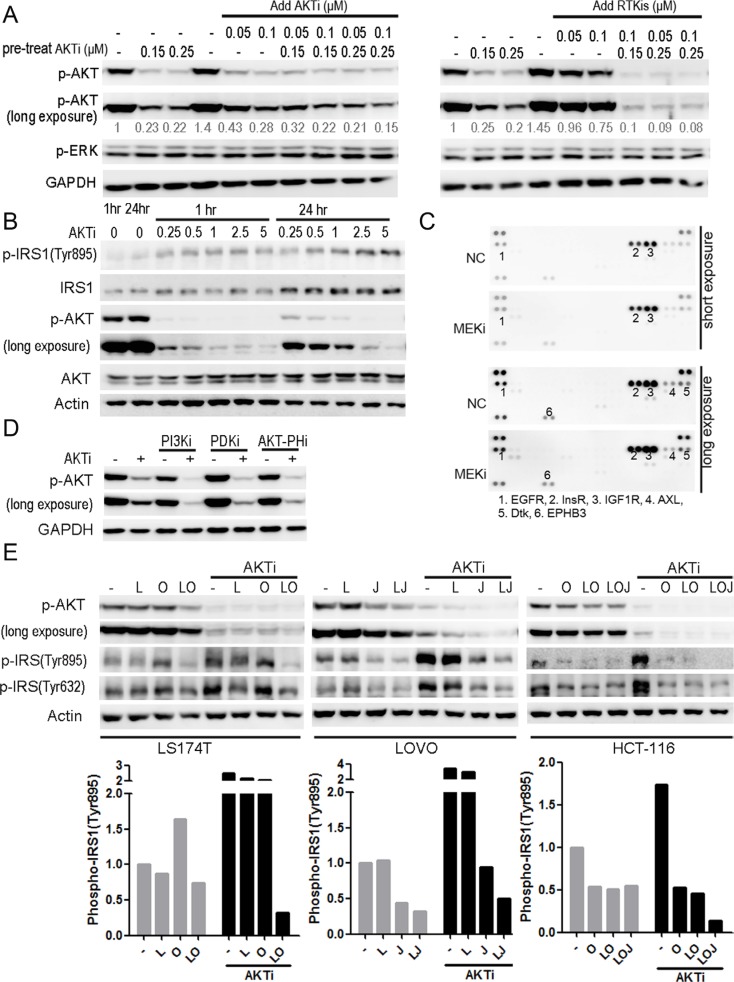
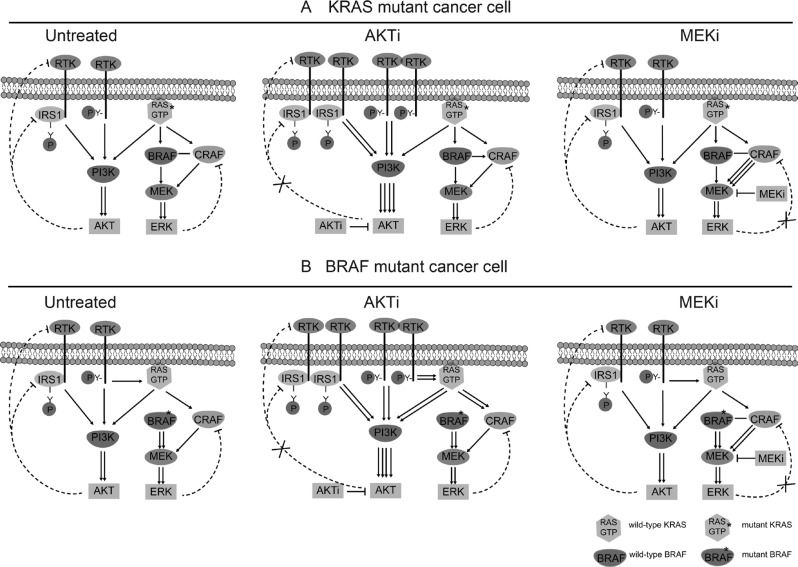
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
