

Stromal Interaction Molecule 1 (STIM1) Regulates ATP-sensitive Potassium (KATP) and Store-operated Ca2+ Channels in MIN6 β-Cells
- PMID: 28003364
- PMCID: PMC5313099
- DOI: 10.1074/jbc.M116.767681
Stromal Interaction Molecule 1 (STIM1) Regulates ATP-sensitive Potassium (KATP) and Store-operated Ca2+ Channels in MIN6 β-Cells
Abstract
Stromal interaction molecule 1 (STIM1) regulates store-operated Ca2+ entry (SOCE) and other ion channels either as an endoplasmic reticulum Ca2+-sensing protein or when present in the plasma membrane. However, the role of STIM1 in insulin-secreting β-cells is unresolved. We report that lowering expression of STIM1, the gene that encodes STIM1, in insulin-secreting MIN6 β-cells with RNA interference inhibits SOCE and ATP-sensitive K+ (KATP) channel activation. The effects of STIM1 knockdown were reversed by transduction of MIN6 cells with an adenovirus gene shuttle vector that expressed human STIM1 Immunoprecipitation studies revealed that STIM1 binds to nucleotide binding fold-1 (NBF1) of the sulfonylurea receptor 1 (SUR1) subunit of the KATP channel. Binding of STIM1 to SUR1 was enhanced by poly-lysine. Our data indicate that SOCE and KATP channel activity are regulated by STIM1. This suggests that STIM1 is a multifunctional signaling effector that participates in the control of membrane excitability and Ca2+ signaling events in β-cells.
Keywords: Ca2+ signaling; KATP channels; STIM1; SUR1; beta cell (B-cell); calcium imaging; cell signaling; electrophysiology; endoplasmic reticulum (ER); store-operated ion channels.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
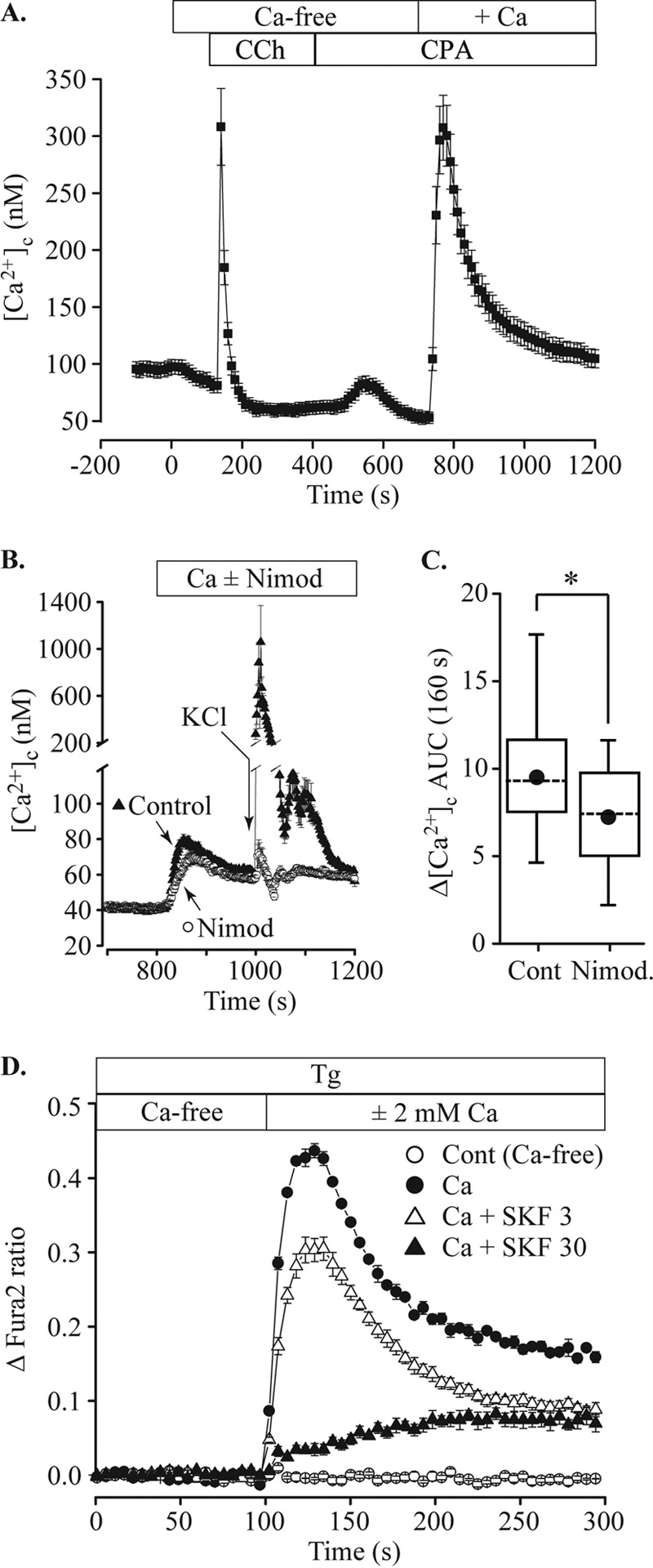
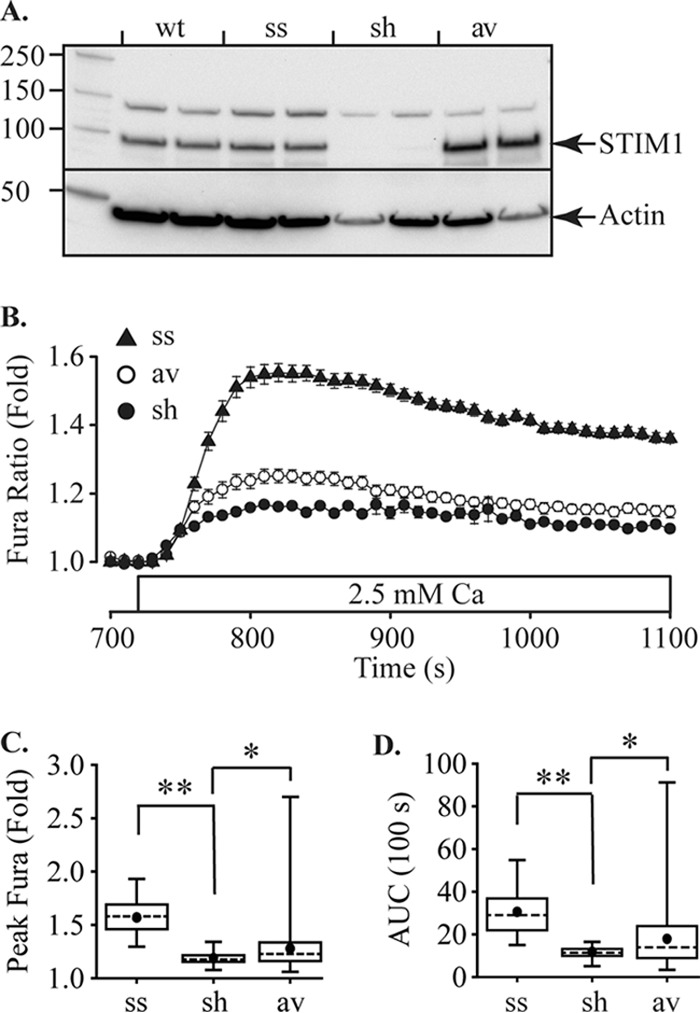
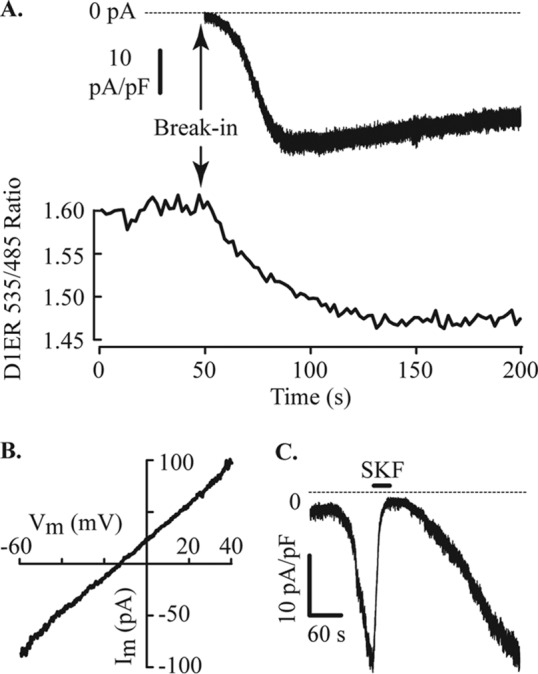
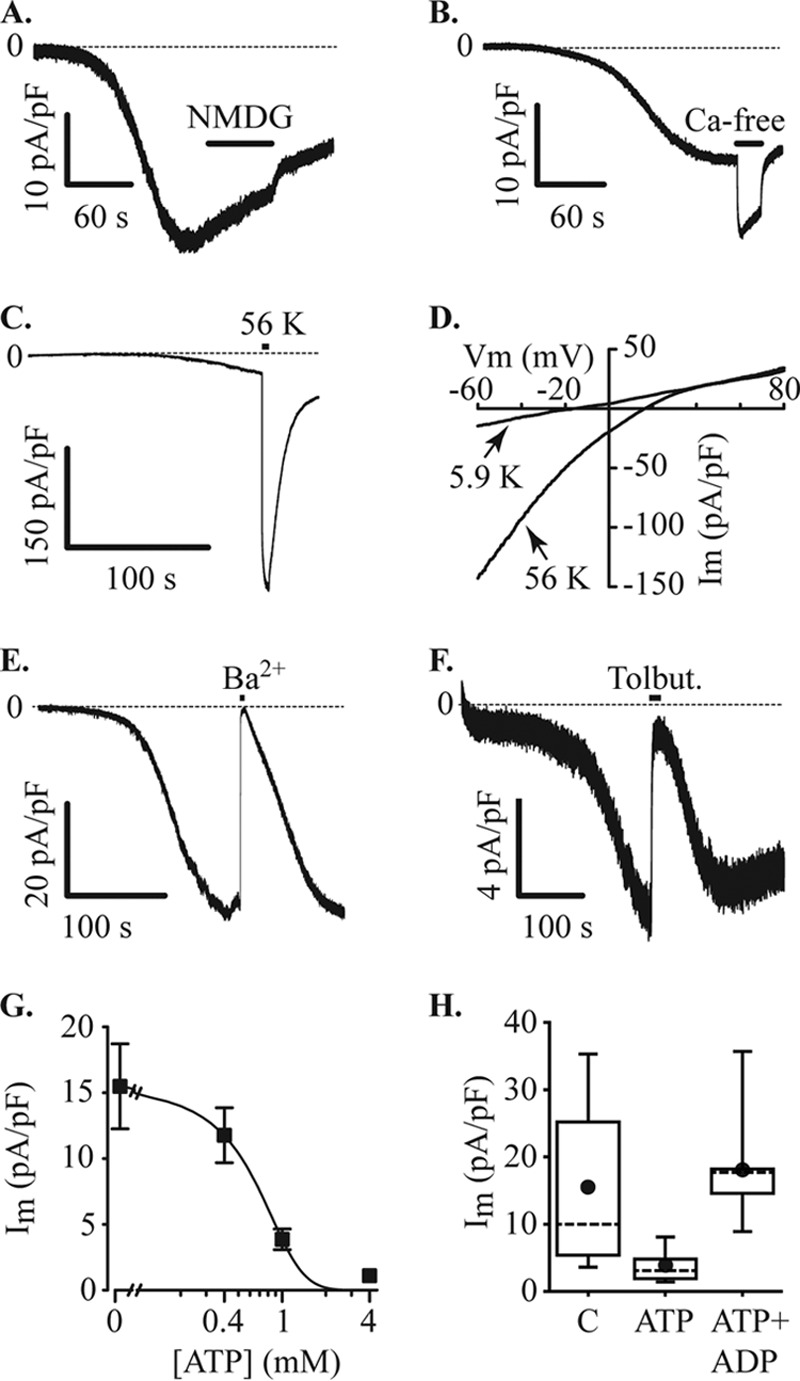
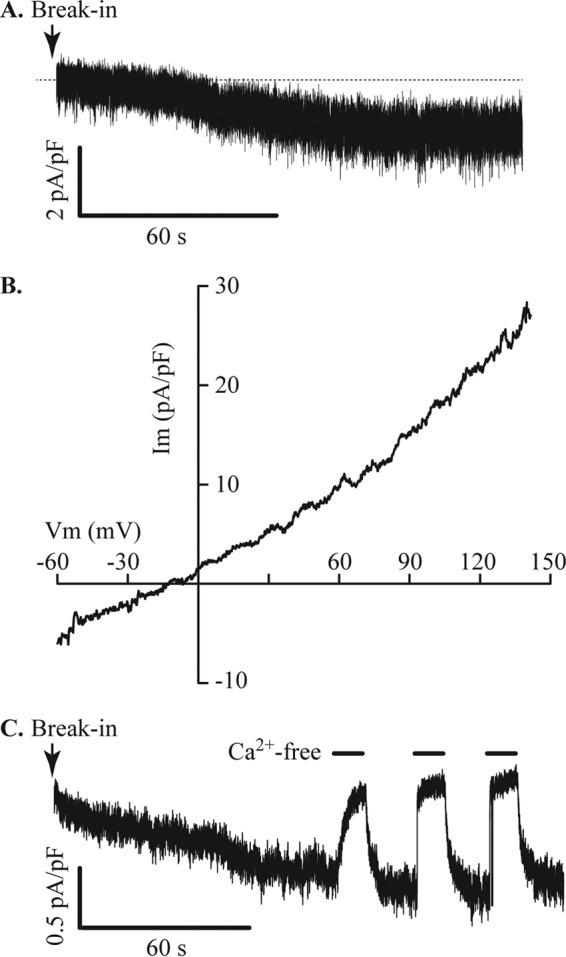
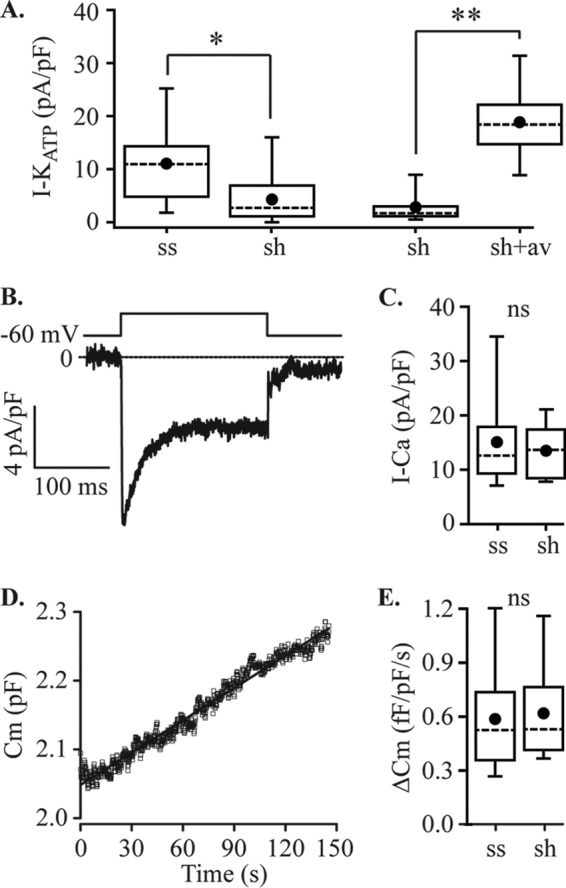
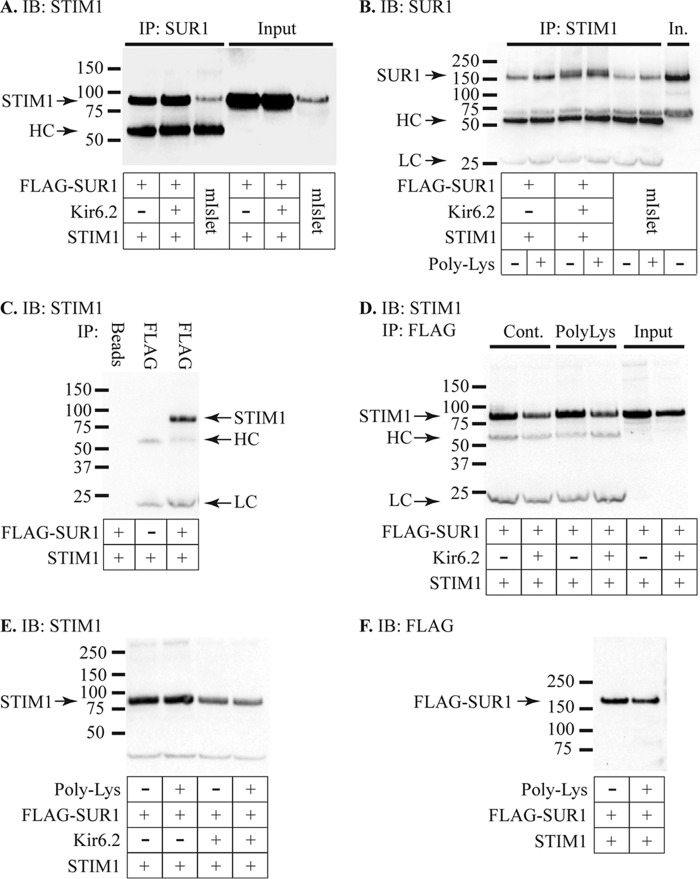
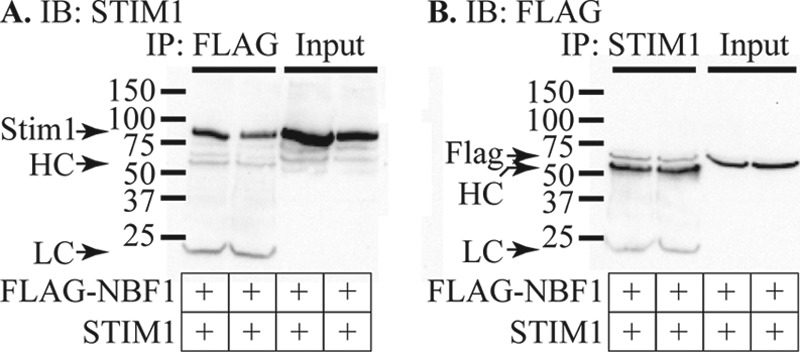
References
-
- Henquin J. C., Nenquin M., Ravier M. A., and Szollosi A. (2009) Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 11, 168–179 - PubMed
-
- Worley J. F. 3rd, McIntyre M. S., Spencer B., and Dukes I. D. (1994) Depletion of intracellular Ca2+ stores activates a maitotoxin-sensitive nonselective cationic current in beta cells. J. Biol. Chem. 269, 32055–32058 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
