

Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes
- PMID: 28003436
- PMCID: PMC5204337
- DOI: 10.1101/gr.201368.115
Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes
Abstract
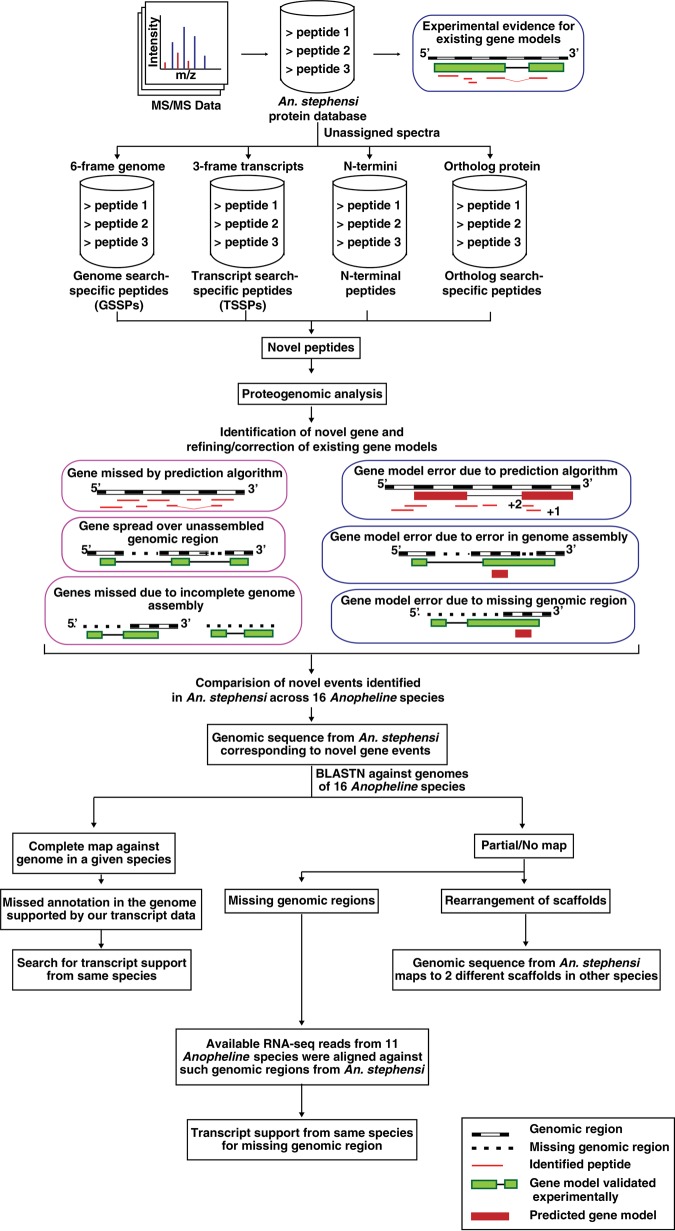
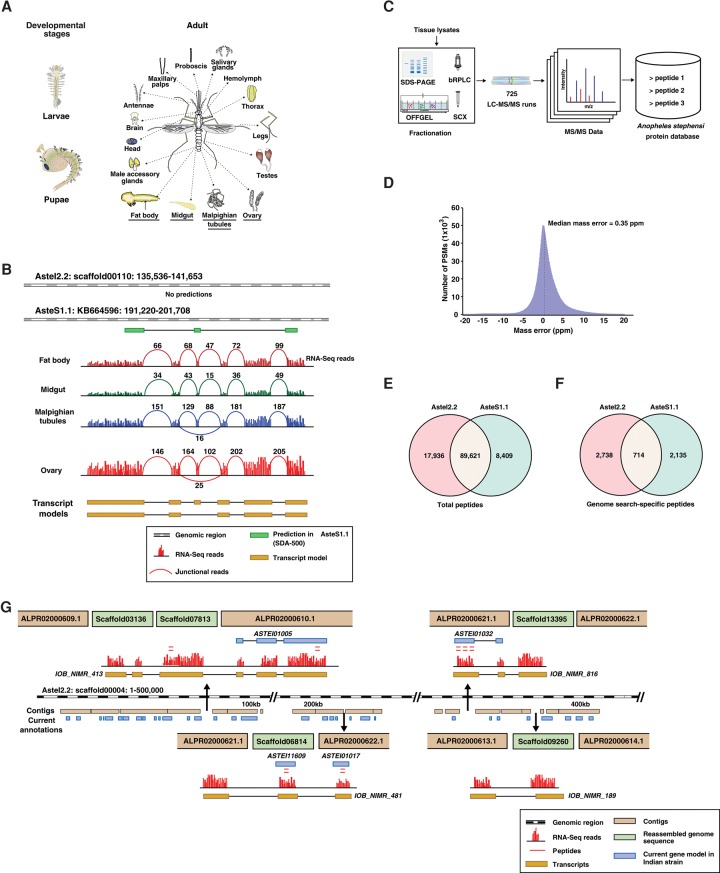
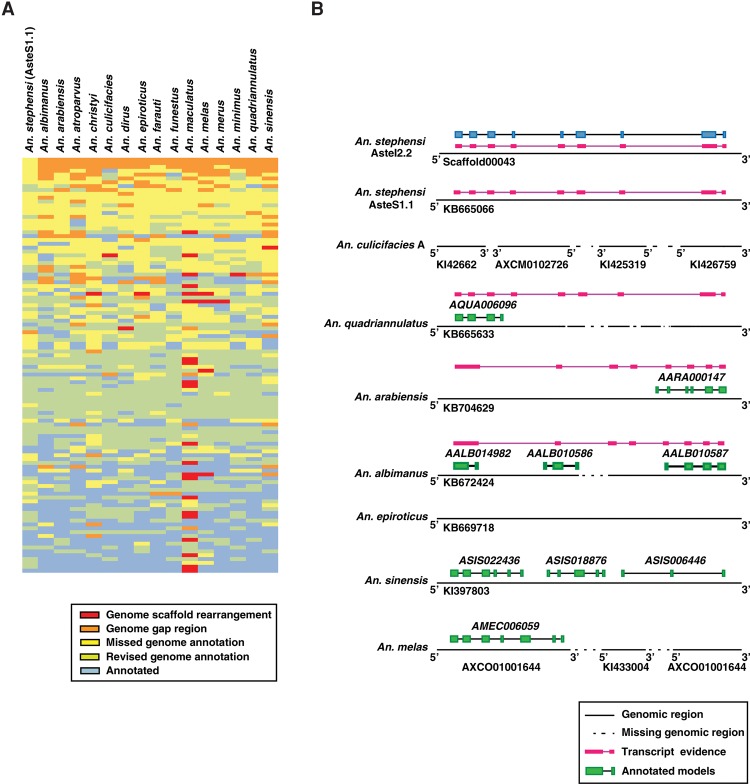
Complementing genome sequence with deep transcriptome and proteome data could enable more accurate assembly and annotation of newly sequenced genomes. Here, we provide a proof-of-concept of an integrated approach for analysis of the genome and proteome of Anopheles stephensi, which is one of the most important vectors of the malaria parasite. To achieve broad coverage of genes, we carried out transcriptome sequencing and deep proteome profiling of multiple anatomically distinct sites. Based on transcriptomic data alone, we identified and corrected 535 events of incomplete genome assembly involving 1196 scaffolds and 868 protein-coding gene models. This proteogenomic approach enabled us to add 365 genes that were missed during genome annotation and identify 917 gene correction events through discovery of 151 novel exons, 297 protein extensions, 231 exon extensions, 192 novel protein start sites, 19 novel translational frames, 28 events of joining of exons, and 76 events of joining of adjacent genes as a single gene. Incorporation of proteomic evidence allowed us to change the designation of more than 87 predicted "noncoding RNAs" to conventional mRNAs coded by protein-coding genes. Importantly, extension of the newly corrected genome assemblies and gene models to 15 other newly assembled Anopheline genomes led to the discovery of a large number of apparent discrepancies in assembly and annotation of these genomes. Our data provide a framework for how future genome sequencing efforts should incorporate transcriptomic and proteomic analysis in combination with simultaneous manual curation to achieve near complete assembly and accurate annotation of genomes.
© 2017 Prasad et al.; Published by Cold Spring Harbor Laboratory Press.
Figures


References
-
- Armengaud J. 2009. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr Opin Microbiol 12: 292–300. - PubMed
-
- Brunner E, Ahrens CH, Mohanty S, Baetschmann H, Loevenich S, Potthast F, Deutsch EW, Panse C, de Lichtenberg U, Rinner O, et al. 2007. A high-quality catalog of the Drosophila melanogaster proteome. Nat Biotechnol 25: 576–583. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources