

Genesis and Dissemination of Highly Pathogenic H5N6 Avian Influenza Viruses
- PMID: 28003485
- PMCID: PMC5309950
- DOI: 10.1128/JVI.02199-16
Genesis and Dissemination of Highly Pathogenic H5N6 Avian Influenza Viruses
Abstract
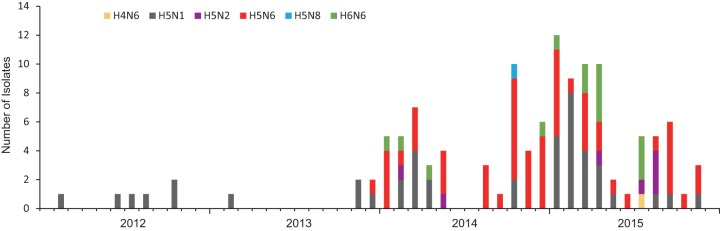
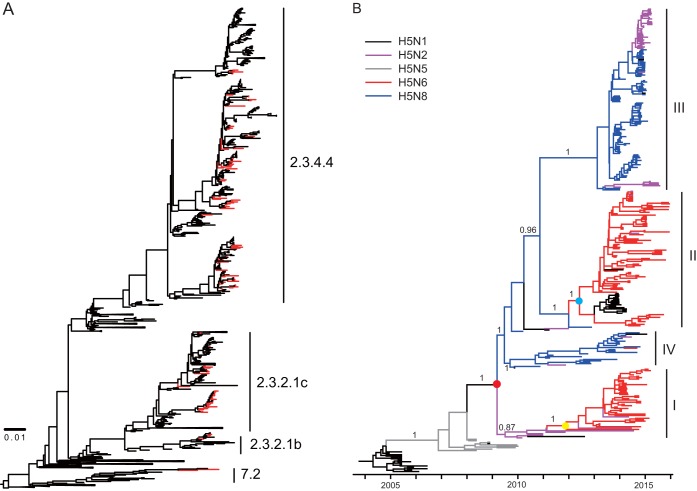
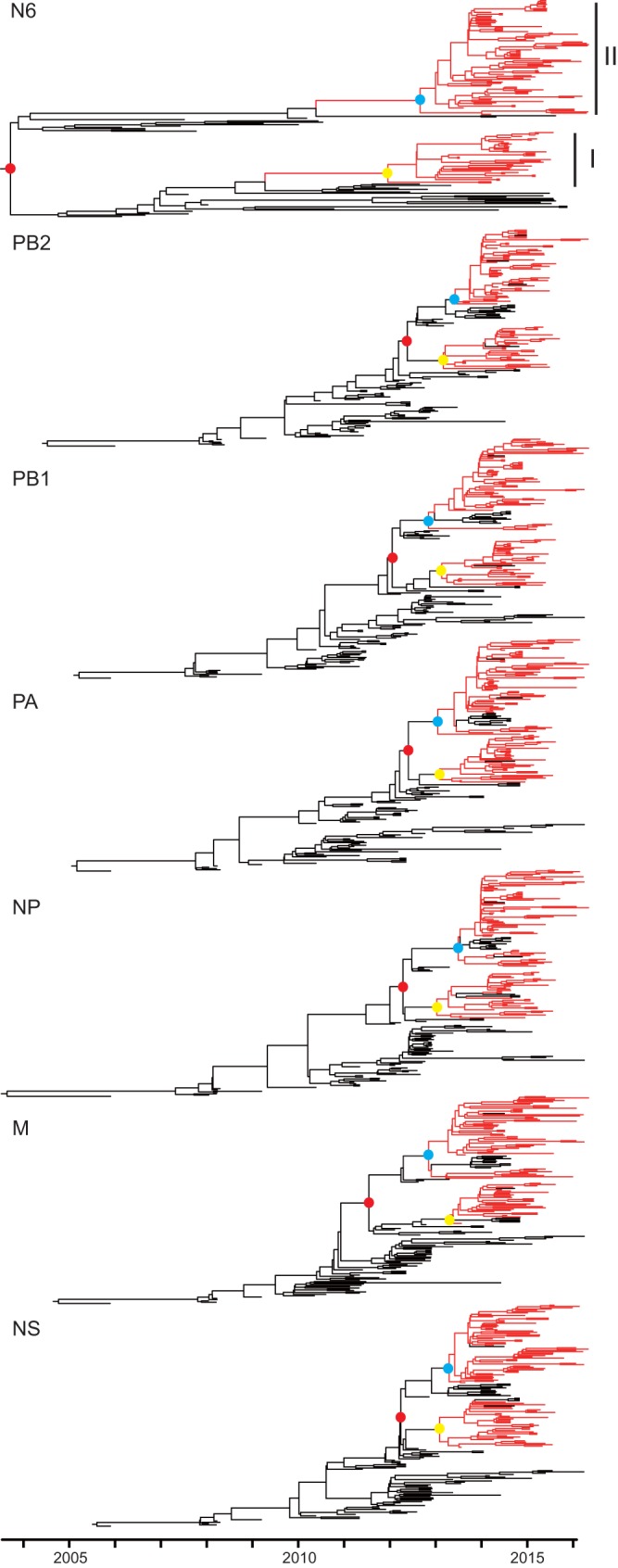
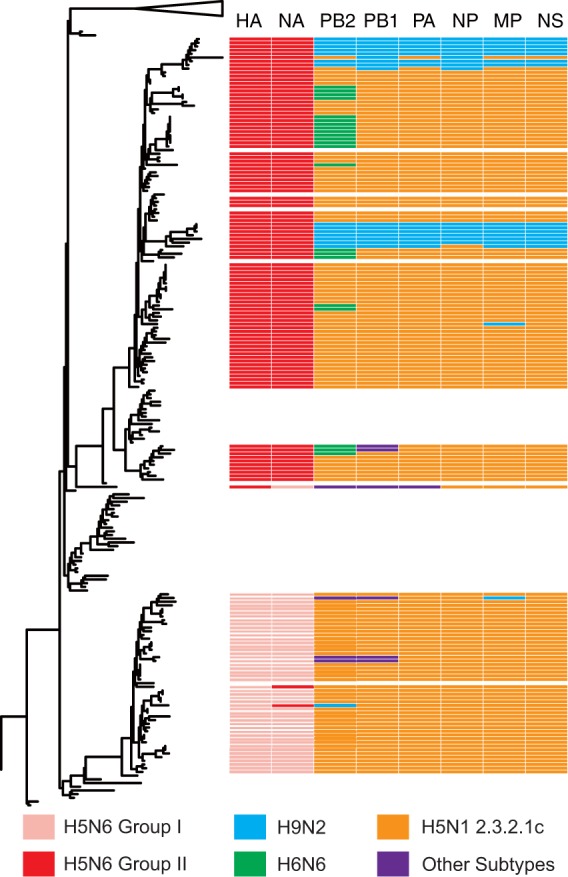
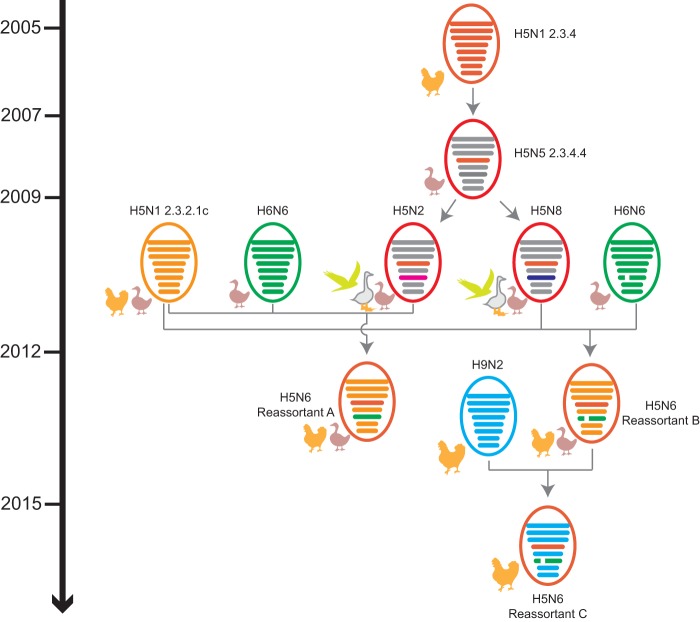
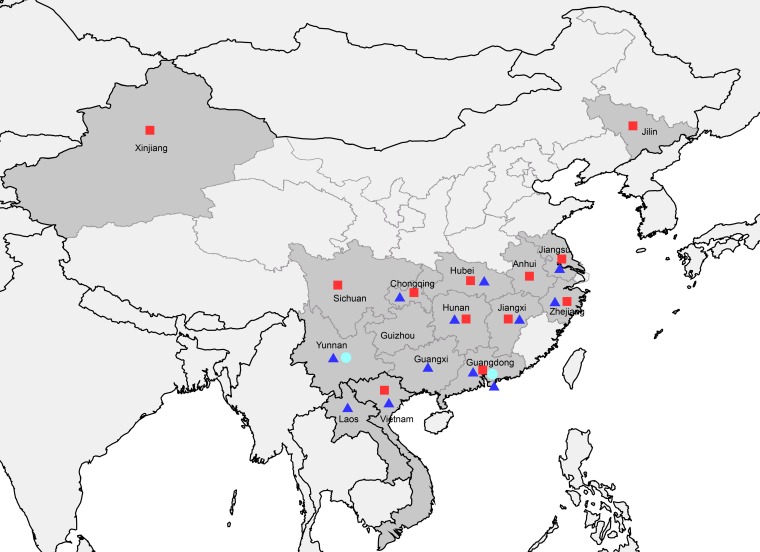
Clade 2.3.4.4 highly pathogenic avian influenza viruses (H5Nx) have spread from Asia to other parts of the world. Since 2014, human infections with clade 2.3.4.4 highly pathogenic avian influenza H5N6 viruses have been continuously reported in China. To investigate the genesis of the virus, we analyzed 123 H5 or N6 environmental viruses sampled from live-poultry markets or farms from 2012 to 2015 in Mainland China. Our results indicated that clade 2.3.4.4 H5N2/N6/N8 viruses shared the same hemagglutinin gene as originated in early 2009. From 2012 to 2015, the genesis of highly pathogenic avian influenza H5N6 viruses occurred via two independent pathways. Three major reassortant H5N6 viruses (reassortants A, B, and C) were generated. Internal genes of reassortant A and B viruses and reassortant C viruses derived from clade 2.3.2.1c H5N1 and H9N2 viruses, respectively. Many mammalian adaption mutations and antigenic variations were detected among the three reassortant viruses. Considering their wide circulation and dynamic reassortment in poultry, we highly recommend close monitoring of the viruses in poultry and humans. IMPORTANCE Since 2014, clade 2.3.4.4 highly pathogenic avian influenza (H5Nx) viruses have caused many outbreaks in both wild and domestic birds globally. Severe human cases with novel H5N6 viruses in this group were also reported in China in 2014 and 2015. To investigate the genesis of the genetic diversity of these H5N6 viruses, we sequenced 123 H5 or N6 environmental viruses sampled from 2012 to 2015 in China. Sequence analysis indicated that three major reassortants of these H5N6 viruses had been generated by two independent evolutionary pathways. The H5N6 reassortant viruses had been detected in most provinces of southern China and neighboring countries. Considering the mammalian adaption mutations and antigenic variation detected, the spread of these viruses should be monitored carefully due to their pandemic potential.
Keywords: H5N6; highly pathogenic avian influenza; reassortment.
Copyright © 2017 American Society for Microbiology.
Figures
References
-
- Duan L, Campitelli L, Fan XH, Leung YH, Vijaykrishna D, Zhang JX, Donatelli I, Delogu M, Li KS, Foni E, Chiapponi C, Wu WL, Kai H, Webster RG, Shortridge KF, Peiris JS, Smith GJ, Chen H, Guan Y. 2007. Characterization of low-pathogenic H5 subtype influenza viruses from Eurasia: implications for the origin of highly pathogenic H5N1 viruses. J Virol 81:7529–7539. doi: 10.1128/JVI.00327-07. - DOI - PMC - PubMed
-
- Duan L, Bahl J, Smith GJ, Wang J, Vijaykrishna D, Zhang LJ, Zhang JX, Li KS, Fan XH, Cheung CL, Huang K, Poon LL, Shortridge KF, Webster RG, Peiris JS, Chen H, Guan Y. 2008. The development and genetic diversity of H5N1 influenza virus in China, 1996–2006. Virology 380:243–254. doi: 10.1016/j.virol.2008.07.038. - DOI - PMC - PubMed
-
- Hill SC, Lee YJ, Song BM, Kang HM, Lee EK, Hanna A, Gilbert M, Brown IH, Pybus OG. 2015. Wild waterfowl migration and domestic duck density shape the epidemiology of highly pathogenic H5N8 influenza in the Republic of Korea. Infect Genet Evol 34:267–277. doi: 10.1016/j.meegid.2015.06.014. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
