

Case Reports
doi: 10.1212/WNL.0000000000003535.
Epub 2016 Dec 21.
Expanding the spectrum of congenital myopathy linked to recessive mutations in SCN4A
Affiliations
- PMID: 28003497
- PMCID: PMC5272967
- DOI: 10.1212/WNL.0000000000003535
Item in Clipboard
Case Reports
Expanding the spectrum of congenital myopathy linked to recessive mutations in SCN4A
Neurology.
.
No abstract available
Figures
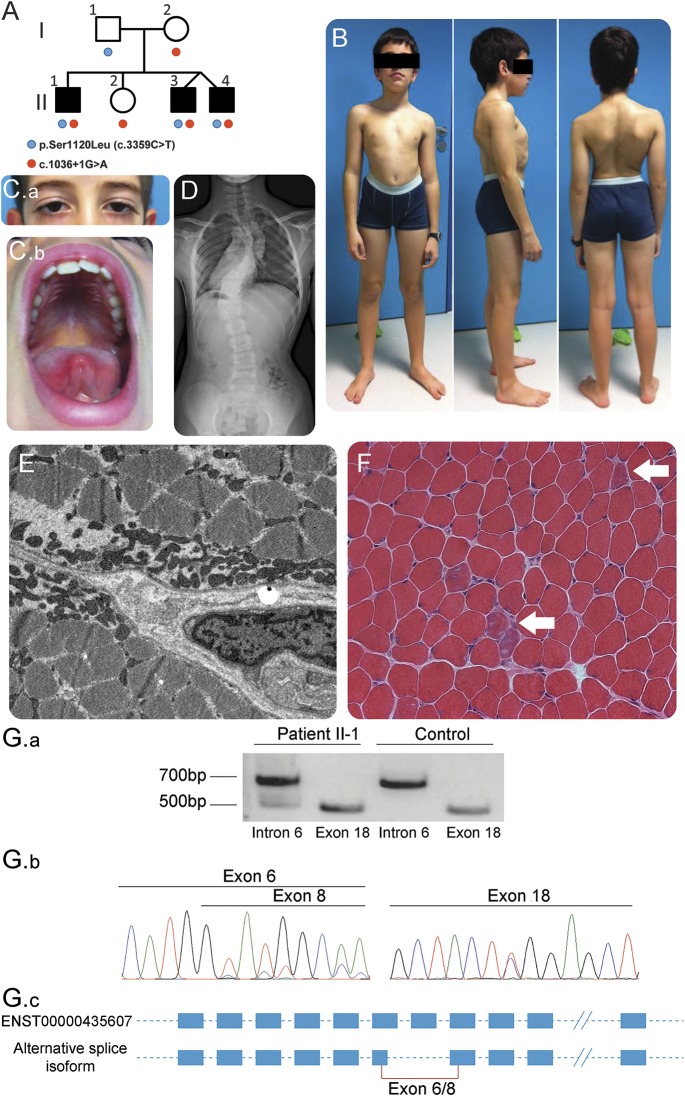
(A) Pedigree of the family with SCN4A mutations indicated compared to reference NM_000334. The 3 affected brothers are represented by shaded symbols. Segregation of the mutations was confirmed by Sanger sequencing. Clinical images of patient II-1: (B) bilateral scapular winging, scoliosis, and lumbar hyperlordosis, (C.a) ptosis in patient II-3, (C.b) high-arched palate in patient II-1. (D) Thoracolumbar spine X-ray shows a severe thoracic scoliosis with double curve in patient II-1 (Cobb angles of 59° for the upper curve and 51° for the lower curve). (E) Muscle biopsy analyses (electron microscopy, ×10,000) presence of an abnormal subsarcolemmal collection of mitochondria with irregular shape and size in patient II-1. (F) Light microscopy (hematoxylin & eosin, ×20) shows the presence of scattered basophilic bona fide regenerating fibers in patient II-2 (white arrows). (G) RNA analysis of patient II-1 muscle. (G.a) Agarose gel electrophoresis of reverse transcription PCR amplification product reveals an additional splice product generated by the mutation in intron 6. (G.b) Chromatopherograms show that the impact of the mutation in intron 6 leads to the use of a cryptic donor site in exon 6 and skipping of exon 7, producing a shorter RNA with an in-frame deletion of 183 nt (predicted 61aa deletion in the third extracellular loop of the first transmembrane domain) (left) and the presence of the mutation in exon 18 affecting a conserved amino acid located in the second extracellular loop of the third transmembrane domain (right). (G.c) Scheme of the new splice product created by the mutation in intron 6.
References
-
- Plassart E, Reboul J, Rime CS, et al. . Mutations in the muscle sodium channel gene (SCN4A) in 13 French families with hyperkalemic periodic paralysis and paramyotonia congenita: phenotype to genotype correlations and demonstration of the predominance of two mutations. Eur J Hum Genet 1994;2:110–124. - PubMed
-
- Ptácek LJ, Tawil R, Griggs RC, et al. . Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology 1994;44:1500–1503. - PubMed
-
- Sternberg D, Maisonobe T, Jurkat-Rott K, et al. . Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001;124:1091–1099. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources