

Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt
- PMID: 28004654
- PMCID: PMC5192182
- DOI: 10.1038/ncomms13817
Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt
Abstract
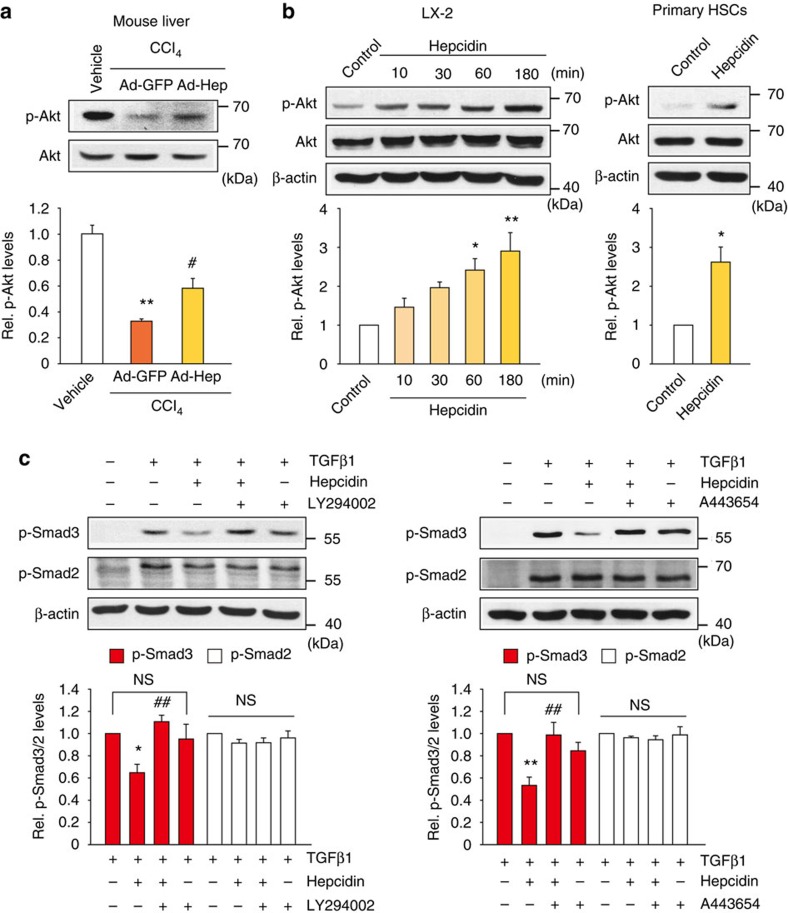
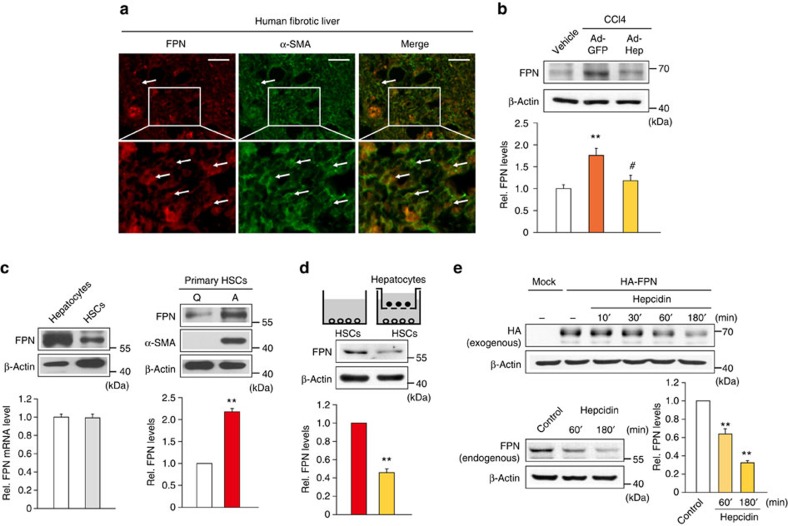
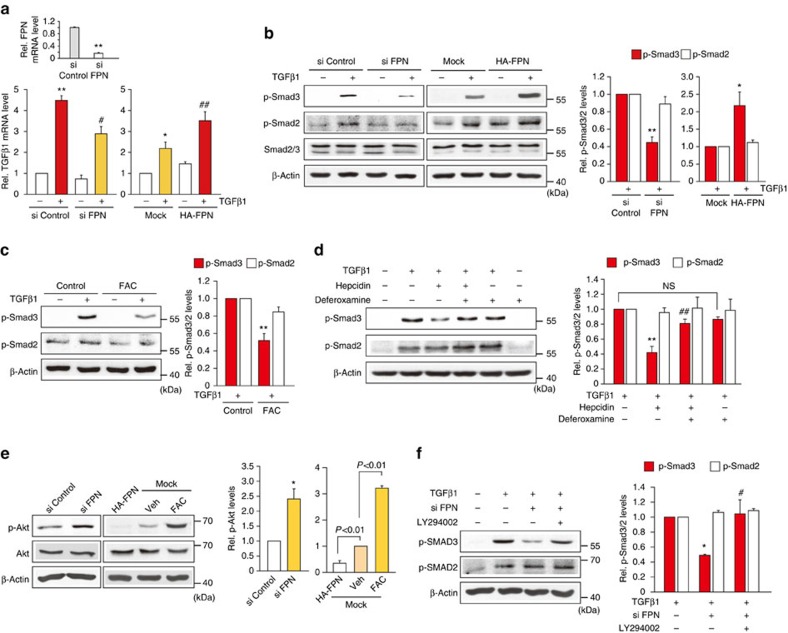
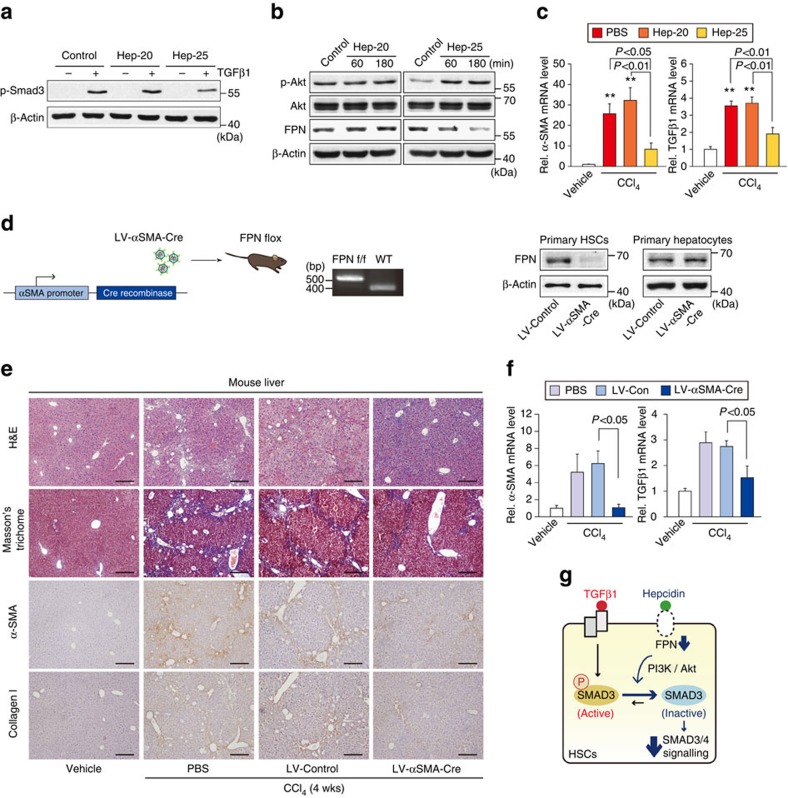
Hepatic stellate cell (HSC) activation on liver injury facilitates fibrosis. Hepatokines affecting HSCs are largely unknown. Here we show that hepcidin inhibits HSC activation and ameliorates liver fibrosis. We observe that hepcidin levels are inversely correlated with exacerbation of fibrosis in patients, and also confirm the relationship in animal models. Adenoviral delivery of hepcidin to mice attenuates liver fibrosis induced by CCl4 treatment or bile duct ligation. In cell-based assays, either hepcidin from hepatocytes or exogenous hepcidin suppresses HSC activation by inhibiting TGFβ1-mediated Smad3 phosphorylation via Akt. In activated HSCs, ferroportin is upregulated, which can be prevented by hepcidin treatment. Similarly, ferroportin knockdown in HSCs prohibits TGFβ1-inducible Smad3 phosphorylation and increases Akt phosphorylation, whereas ferroportin over-expression has the opposite effect. HSC-specific ferroportin deletion also ameliorates liver fibrosis. In summary, hepcidin suppresses liver fibrosis by impeding TGFβ1-induced Smad3 phosphorylation in HSCs, which depends on Akt activated by a deficiency of ferroportin.
Conflict of interest statement
S.R. is a consultant for Isis, Bayer A.G., Merganser Biotech and Novartis Pharmaceuticals. He also holds an equity/ownership interest in Merganser Biotech and is a co-inventor for patents US8058061 B2 C12N 20111115 and US7541179 B2 C12N 20090602 about the generation of lentiviral vectors for the cure of hemoglobinopathies. The remaining authors declare no competing interests.
Figures





References
-
- Mabuchi A. et al. Role of hepatic stellate cell/hepatocyte interaction and activation of hepatic stellate cells in the early phase of liver regeneration in the rat. J. Hepatol. 40, 910–916 (2004). - PubMed
-
- Sawitza I., Kordes C., Reister S. & Haussinger D. The niche of stellate cells within rat liver. Hepatology 50, 1617–1624 (2009). - PubMed
-
- Hernandez-Gea V. & Friedman S. L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6, 425–456 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
