

Novel Candidate Genes and a Wide Spectrum of Structural and Point Mutations Responsible for Inherited Retinal Dystrophies Revealed by Exome Sequencing
- PMID: 28005958
- PMCID: PMC5179108
- DOI: 10.1371/journal.pone.0168966
Novel Candidate Genes and a Wide Spectrum of Structural and Point Mutations Responsible for Inherited Retinal Dystrophies Revealed by Exome Sequencing
Abstract
Background: NGS-based genetic diagnosis has completely revolutionized the human genetics field. In this study, we have aimed to identify new genes and mutations by Whole Exome Sequencing (WES) responsible for inherited retinal dystrophies (IRD).
Methods: A cohort of 33 pedigrees affected with a variety of retinal disorders was analysed by WES. Initial prioritization analysis included around 300 IRD-associated genes. In non-diagnosed families a search for pathogenic mutations in novel genes was undertaken.
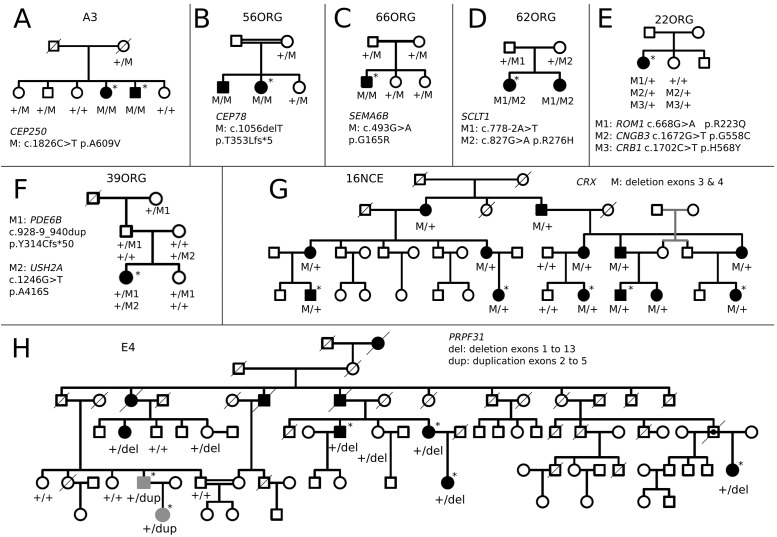
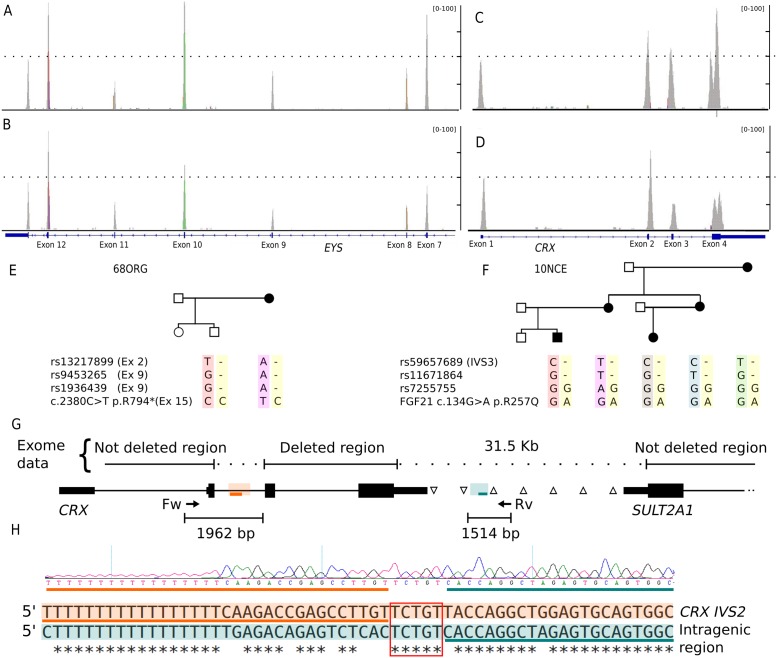
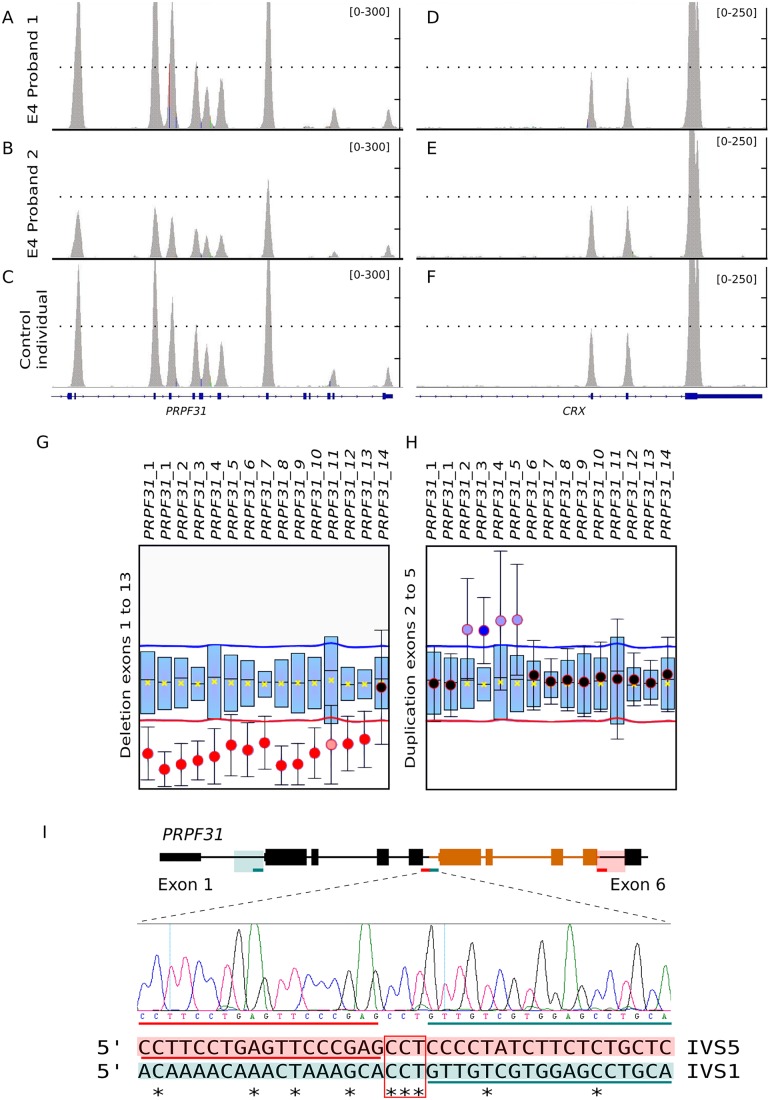
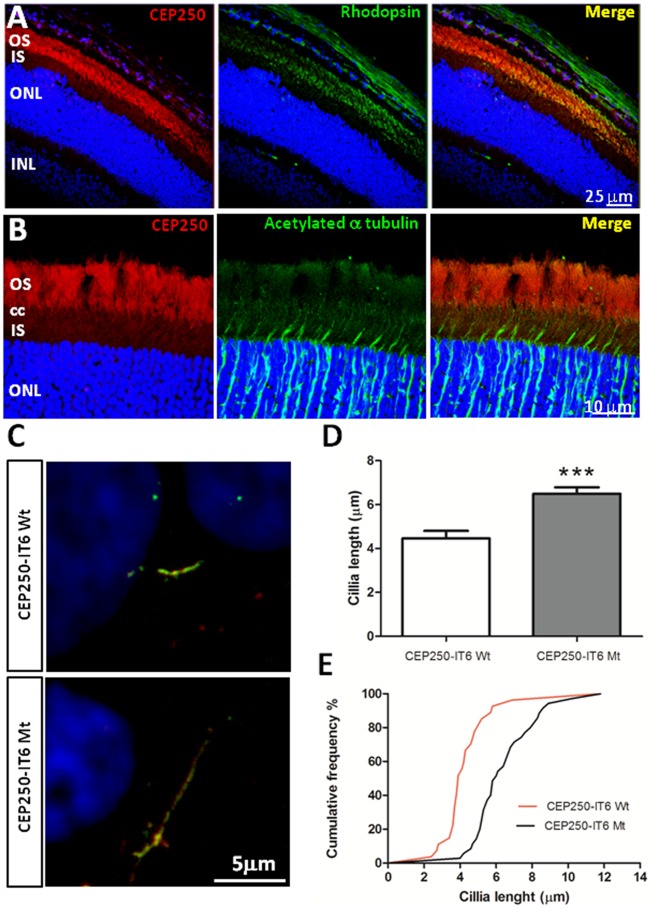
Results: Genetic diagnosis was attained in 18 families. Moreover, a plausible candidate is proposed for 10 more cases. Two thirds of the mutations were novel, including 4 chromosomal rearrangements, which expand the IRD allelic heterogeneity and highlight the contribution of private mutations. Our results prompted clinical re-evaluation of some patients resulting in assignment to a syndromic instead of non-syndromic IRD. Notably, WES unveiled four new candidates for non-syndromic IRD: SEMA6B, CEP78, CEP250, SCLT1, the two latter previously associated to syndromic disorders. We provide functional data supporting that missense mutations in CEP250 alter cilia formation.
Conclusion: The diagnostic efficiency of WES, and strictly following the ACMG/AMP criteria is 55% in reported causative genes or functionally supported new candidates, plus 30% families in which likely pathogenic or VGUS/VUS variants were identified in plausible candidates. Our results highlight the clinical utility of WES for molecular diagnosis of IRD, provide a wider spectrum of mutations and concomitant genetic variants, and challenge our view on syndromic vs non-syndromic, and causative vs modifier genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Identification of Novel and Recurrent Disease-Causing Mutations in Retinal Dystrophies Using Whole Exome Sequencing (WES): Benefits and Limitations.PLoS One. 2016 Jul 8;11(7):e0158692. doi: 10.1371/journal.pone.0158692. eCollection 2016. PLoS One. 2016. PMID: 27391102 Free PMC article.
-
[Comparison study of whole exome sequencing and targeted panel sequencing in molecular diagnosis of inherited retinal dystrophies].Beijing Da Xue Xue Bao Yi Xue Ban. 2020 Oct 18;52(5):836-844. doi: 10.19723/j.issn.1671-167X.2020.05.007. Beijing Da Xue Xue Bao Yi Xue Ban. 2020. PMID: 33047716 Free PMC article. Chinese.
-
Scaling New Heights in the Genetic Diagnosis of Inherited Retinal Dystrophies.Adv Exp Med Biol. 2019;1185:215-219. doi: 10.1007/978-3-030-27378-1_35. Adv Exp Med Biol. 2019. PMID: 31884614 Review.
-
Deciphering the genetic architecture and ethnographic distribution of IRD in three ethnic populations by whole genome sequence analysis.PLoS Genet. 2021 Oct 18;17(10):e1009848. doi: 10.1371/journal.pgen.1009848. eCollection 2021 Oct. PLoS Genet. 2021. PMID: 34662339 Free PMC article.
-
Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies.Orphanet J Rare Dis. 2022 May 12;17(1):197. doi: 10.1186/s13023-022-02340-7. Orphanet J Rare Dis. 2022. PMID: 35551639 Free PMC article. Review.
Cited by
-
Long-Read Sequencing to Unravel Complex Structural Variants of CEP78 Leading to Cone-Rod Dystrophy and Hearing Loss.Front Cell Dev Biol. 2021 Apr 21;9:664317. doi: 10.3389/fcell.2021.664317. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33968938 Free PMC article.
-
The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification.Hum Genet. 2022 Apr;141(3-4):709-735. doi: 10.1007/s00439-022-02448-7. Epub 2022 Mar 30. Hum Genet. 2022. PMID: 35353227 Free PMC article. Review.
-
CEP78 functions downstream of CEP350 to control biogenesis of primary cilia by negatively regulating CP110 levels.Elife. 2021 Jul 14;10:e63731. doi: 10.7554/eLife.63731. Elife. 2021. PMID: 34259627 Free PMC article.
-
High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative.Sci Rep. 2018 Nov 20;8(1):17113. doi: 10.1038/s41598-018-35085-0. Sci Rep. 2018. PMID: 30459346 Free PMC article.
-
Prevalence of mutations in inherited retinal diseases: A comparison between the United States and India.Mol Genet Genomic Med. 2020 Feb;8(2):e1081. doi: 10.1002/mgg3.1081. Epub 2019 Dec 9. Mol Genet Genomic Med. 2020. PMID: 31816670 Free PMC article.
References
-
- Marfany G, Gonzàlez-Duarte R. Clinical applications of high-throughput genetic diagnosis in retinal dystrophies: present challenges and future directions. World J Med Genet. 2015;5: 14–22.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
