

Augmentation of S-Nitrosoglutathione Controls Cigarette Smoke-Induced Inflammatory-Oxidative Stress and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis by Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function
- PMID: 28006950
- PMCID: PMC5564030
- DOI: 10.1089/ars.2016.6895
Augmentation of S-Nitrosoglutathione Controls Cigarette Smoke-Induced Inflammatory-Oxidative Stress and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis by Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function
Abstract
Aims: Cigarette smoke (CS)-mediated acquired cystic fibrosis transmembrane conductance regulator (CFTR)-dysfunction, autophagy-impairment, and resulting inflammatory-oxidative/nitrosative stress leads to chronic obstructive pulmonary disease (COPD)-emphysema pathogenesis. Moreover, nitric oxide (NO) signaling regulates lung function decline, and low serum NO levels that correlates with COPD severity. Hence, we aim to evaluate here the effects and mechanism(s) of S-nitrosoglutathione (GSNO) augmentation in regulating inflammatory-oxidative stress and COPD-emphysema pathogenesis.
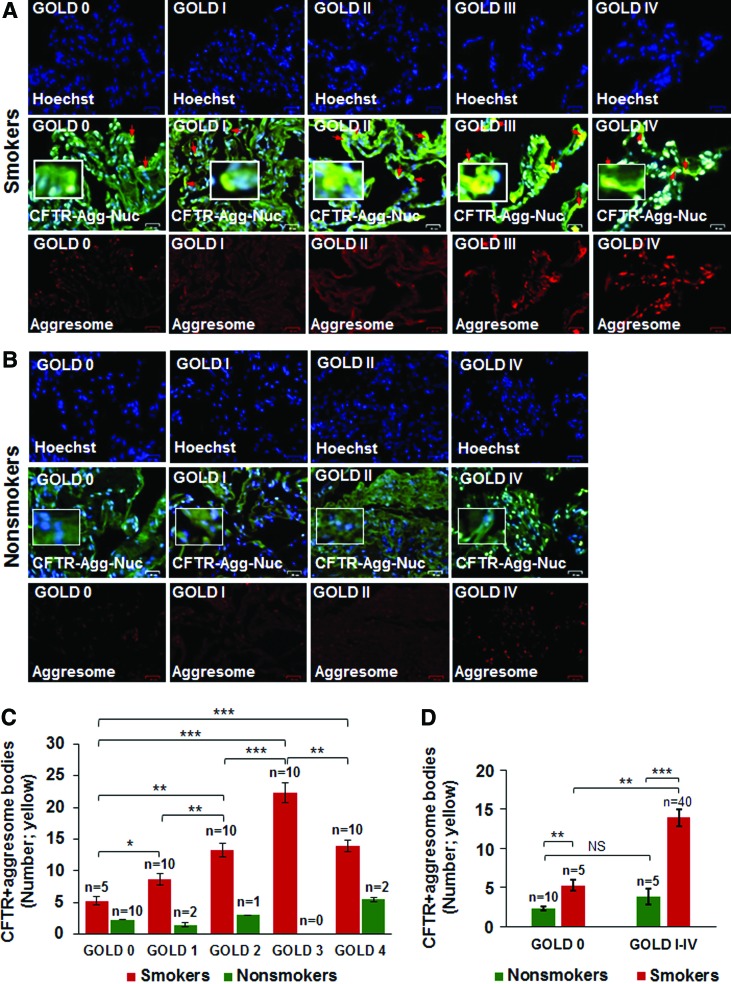
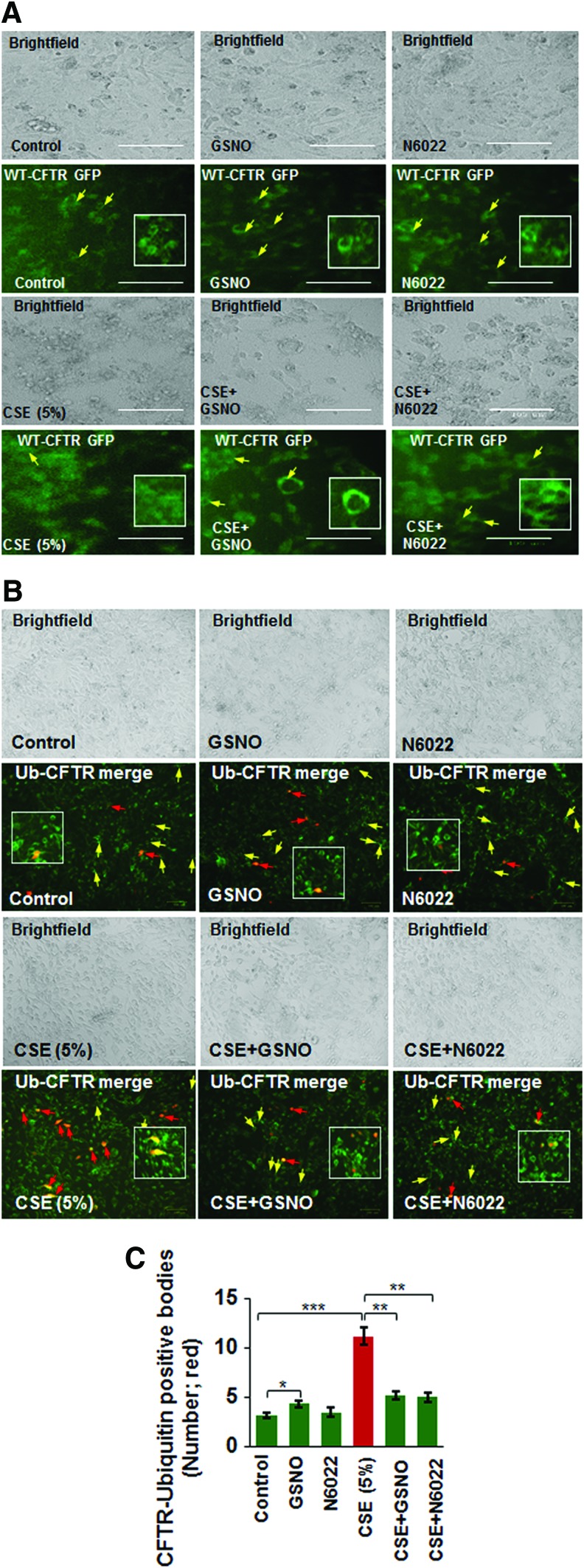
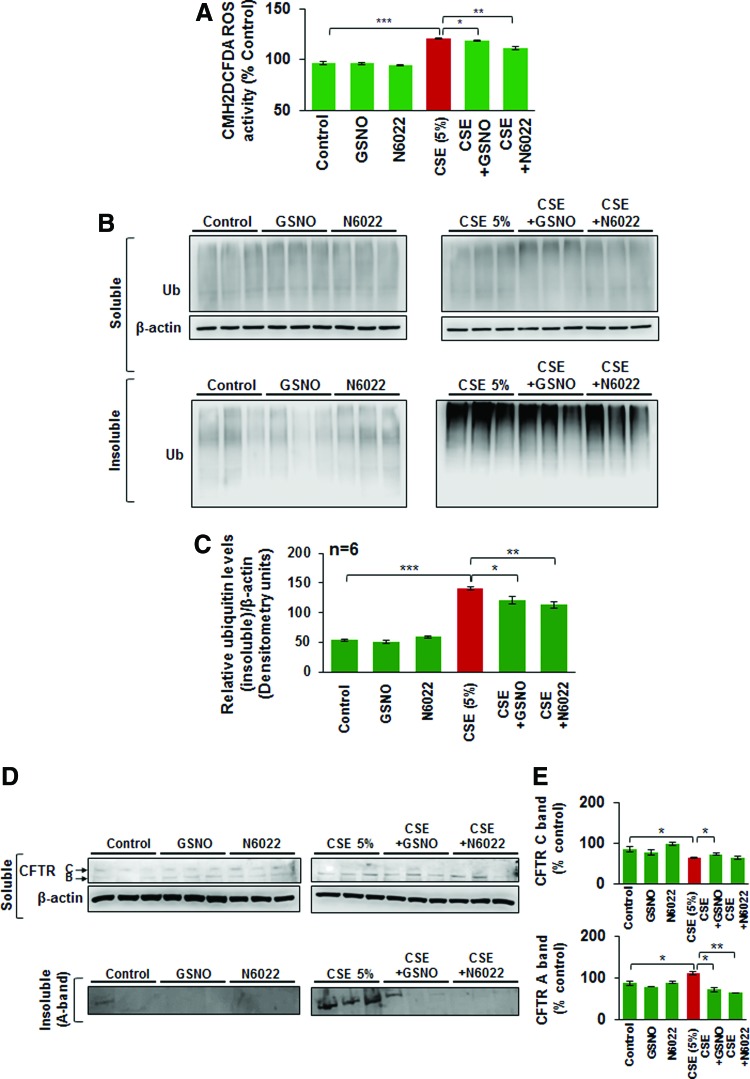
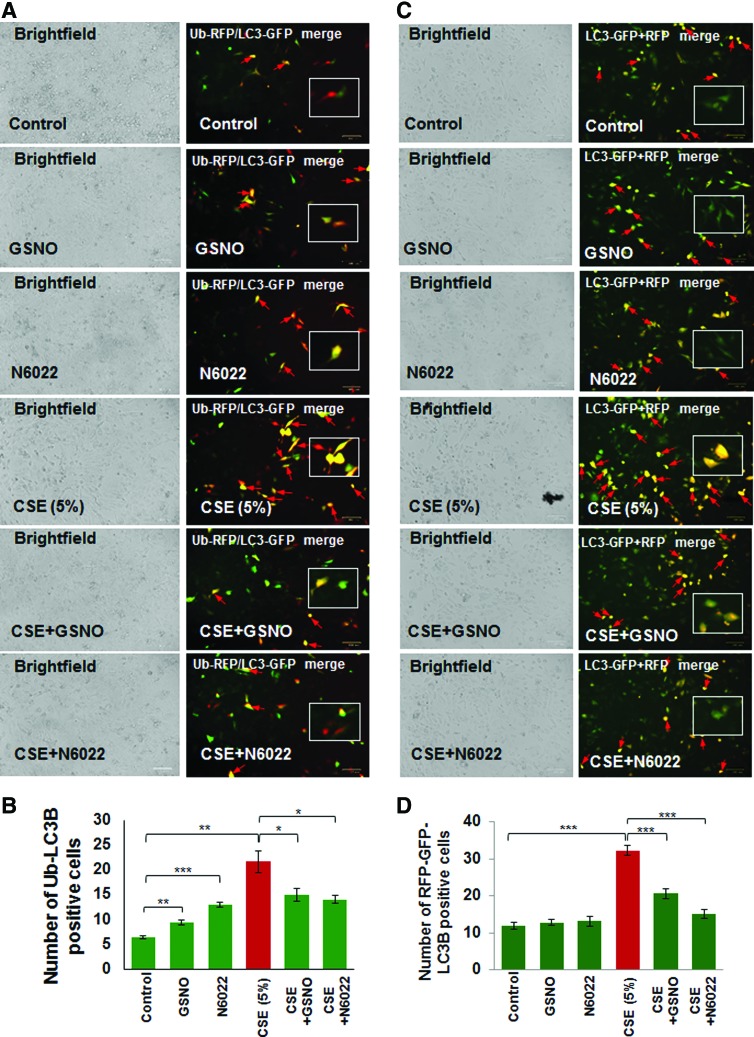
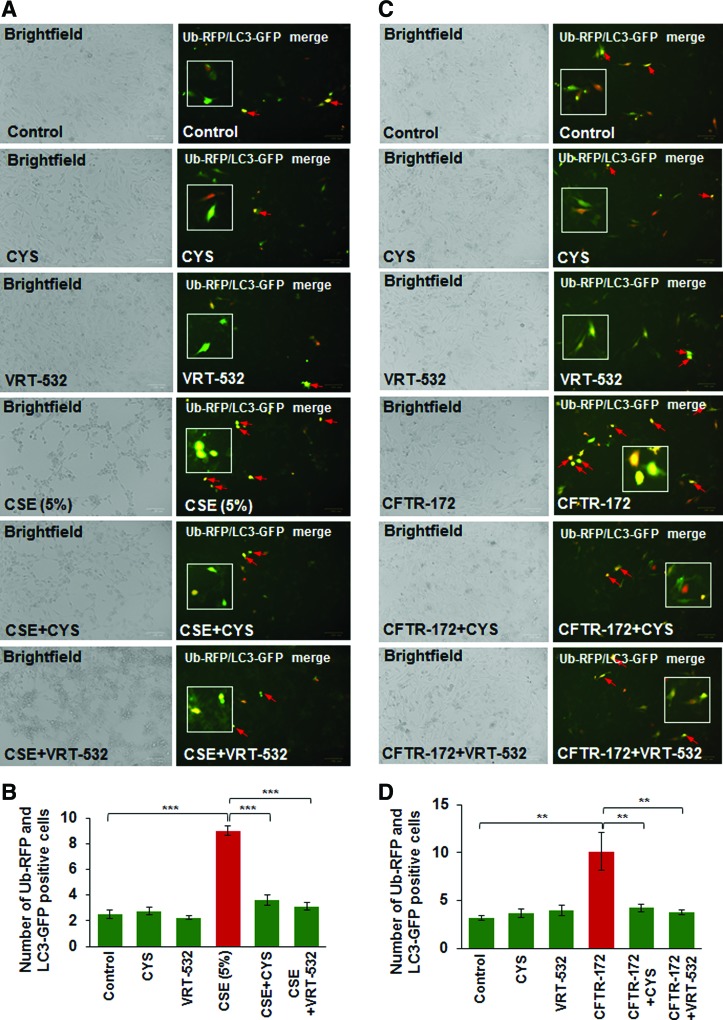
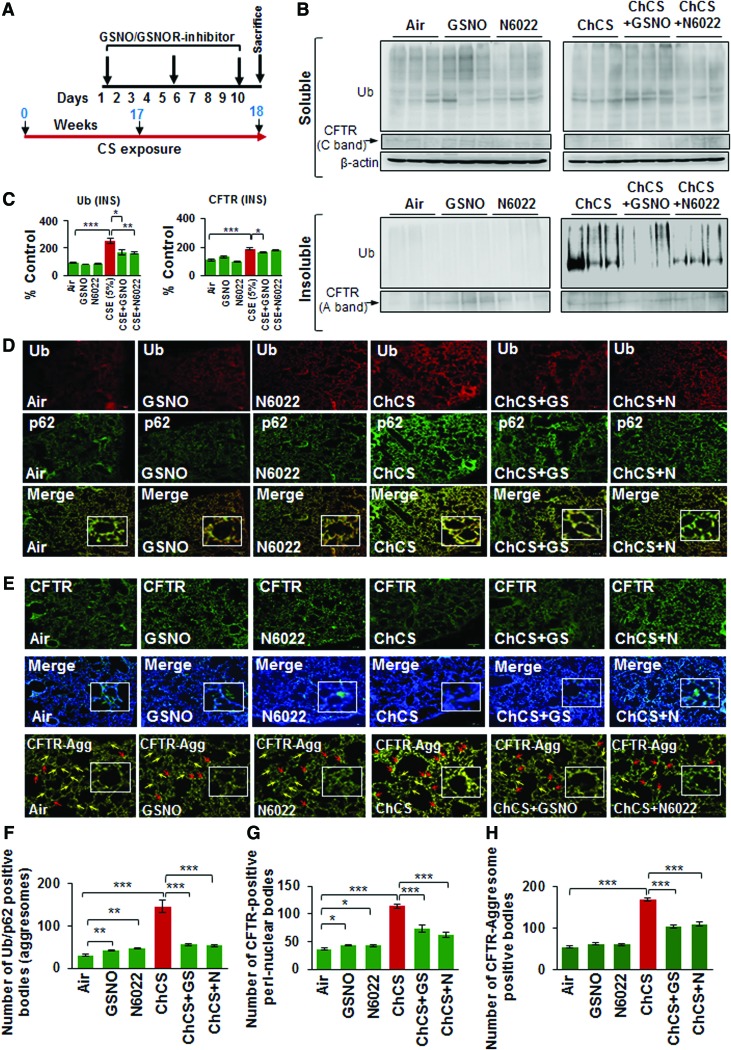
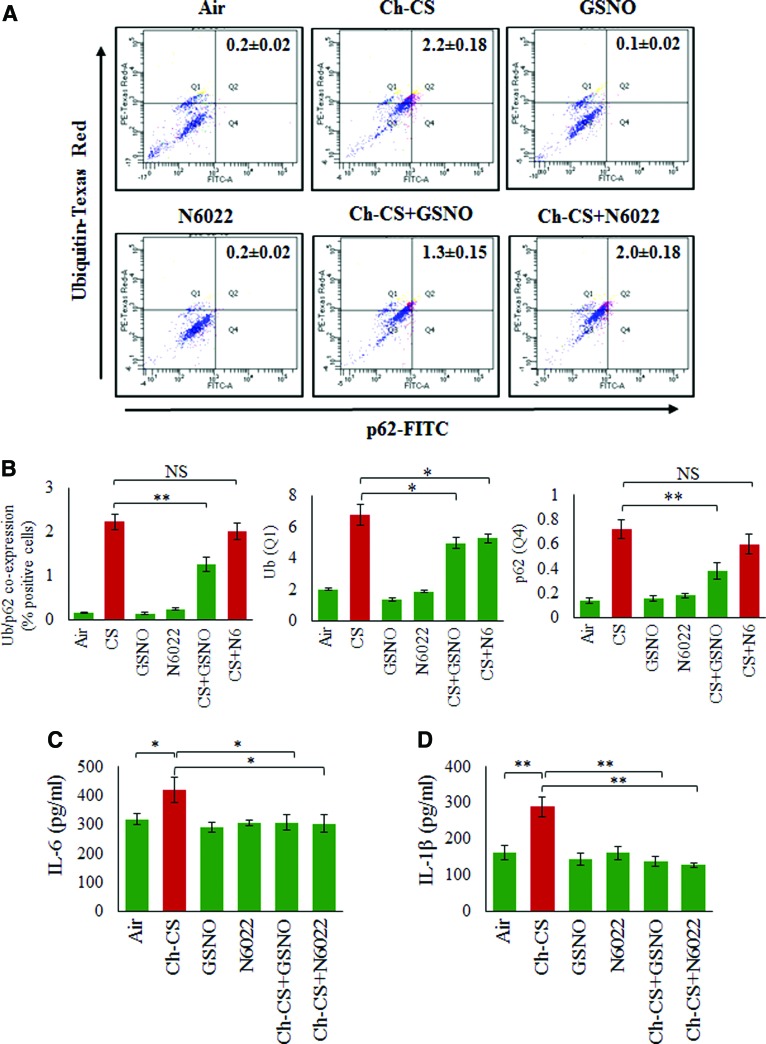
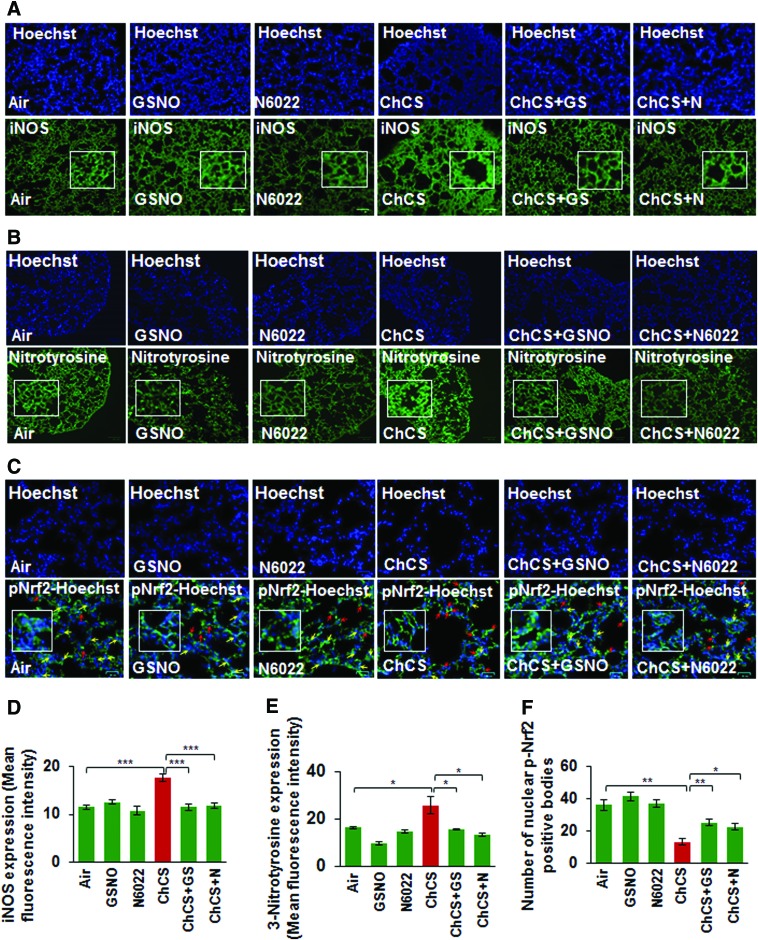
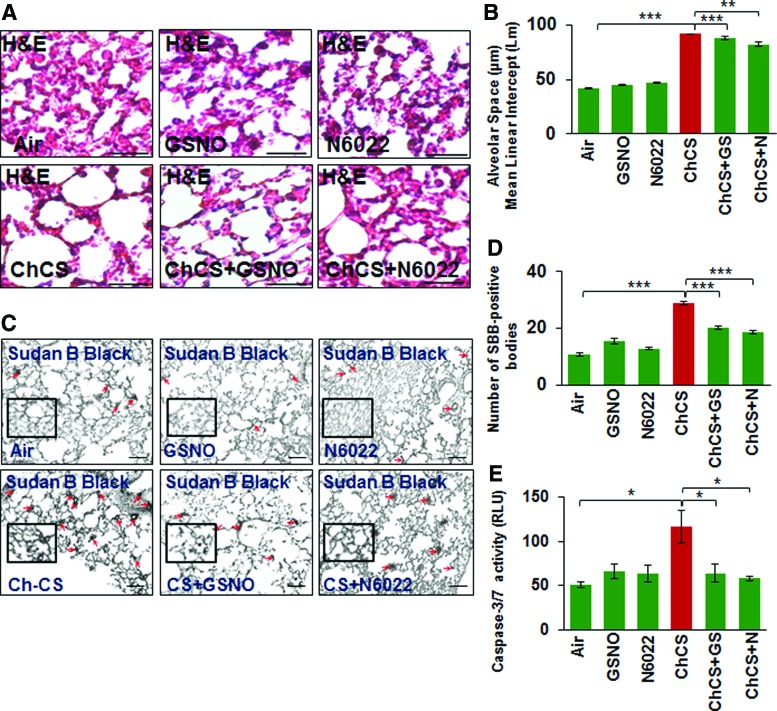
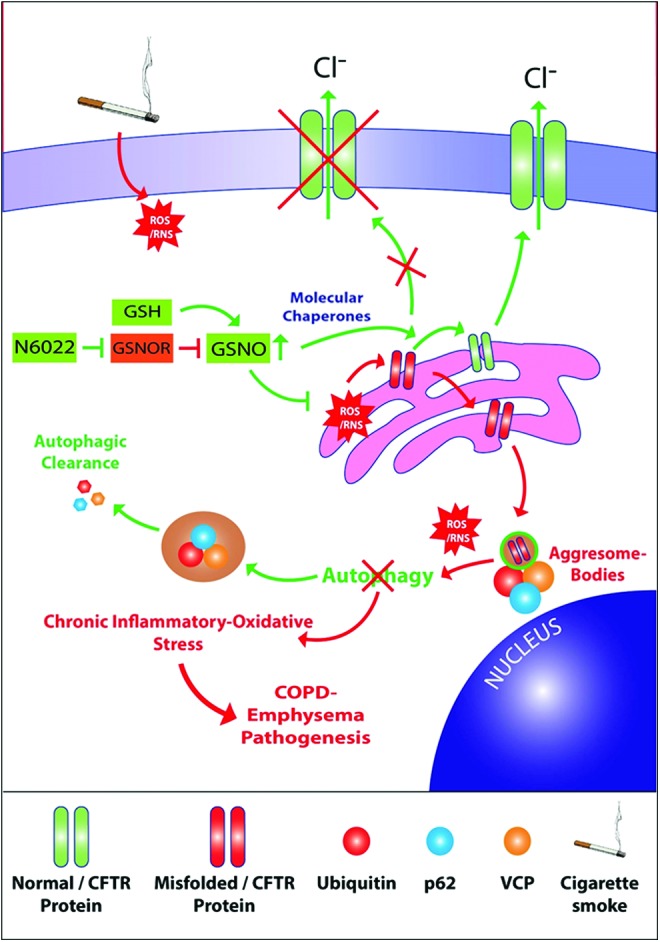
Results: Our data shows that cystic fibrosis transmembrane conductance regulator (CFTR) colocalizes with aggresome bodies in the lungs of COPD subjects with increasing emphysema severity (Global Initiative for Chronic Obstructive Lung Disease [GOLD] I - IV) compared to nonemphysema controls (GOLD 0). We further demonstrate that treatment with GSNO or S-nitrosoglutathione reductase (GSNOR)-inhibitor (N6022) significantly inhibits cigarette smoke extract (CSE; 5%)-induced decrease in membrane CFTR expression by rescuing it from ubiquitin (Ub)-positive aggresome bodies (p < 0.05). Moreover, GSNO restoration significantly (p < 0.05) decreases CSE-induced reactive oxygen species (ROS) activation and autophagy impairment (decreased accumulation of ubiquitinated proteins in the insoluble protein fractions and restoration of autophagy flux). In addition, GSNO augmentation inhibits protein misfolding as CSE-induced colocalization of ubiquitinated proteins and LC3B (in autophagy bodies) is significantly reduced by GSNO/N6022 treatment. We verified using the preclinical COPD-emphysema murine model that chronic CS (Ch-CS)-induced inflammation (interleukin [IL]-6/IL-1β levels), aggresome formation (perinuclear coexpression/colocalization of ubiquitinated proteins [Ub] and p62 [impaired autophagy marker], and CFTR), oxidative/nitrosative stress (p-Nrf2, inducible nitric oxide synthase [iNOS], and 3-nitrotyrosine expression), apoptosis (caspase-3/7 activity), and alveolar airspace enlargement (Lm) are significantly (p < 0.05) alleviated by augmenting airway GSNO levels. As a proof of concept, we demonstrate that GSNO augmentation suppresses Ch-CS-induced perinuclear CFTR protein accumulation (p < 0.05), which restores both acquired CFTR dysfunction and autophagy impairment, seen in COPD-emphysema subjects.
Innovation: GSNO augmentation alleviates CS-induced acquired CFTR dysfunction and resulting autophagy impairment.
Conclusion: Overall, we found that augmenting GSNO levels controls COPD-emphysema pathogenesis by reducing CS-induced acquired CFTR dysfunction and resulting autophagy impairment and chronic inflammatory-oxidative stress. Antioxid. Redox Signal. 27, 433-451.
Keywords: CFTR; COPD; GSNO; GSNOR; NO; emphysema.
Conflict of interest statement
No competing financial interests exist.
Figures
Similar articles
-
Cigarette Smoke-Induced Acquired Dysfunction of Cystic Fibrosis Transmembrane Conductance Regulator in the Pathogenesis of Chronic Obstructive Pulmonary Disease.Oxid Med Cell Longev. 2018 Apr 23;2018:6567578. doi: 10.1155/2018/6567578. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 29849907 Free PMC article. Review.
-
Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis.Antioxid Redox Signal. 2017 Jul 20;27(3):150-167. doi: 10.1089/ars.2016.6842. Epub 2017 Feb 1. Antioxid Redox Signal. 2017. PMID: 27835930 Free PMC article.
-
Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis.Am J Respir Cell Mol Biol. 2015 Aug;53(2):159-73. doi: 10.1165/rcmb.2014-0107OC. Am J Respir Cell Mol Biol. 2015. PMID: 25490051 Free PMC article.
-
Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis.Free Radic Biol Med. 2019 Feb 1;131:81-97. doi: 10.1016/j.freeradbiomed.2018.11.023. Epub 2018 Nov 28. Free Radic Biol Med. 2019. PMID: 30500419
-
Nano-based rescue of dysfunctional autophagy in chronic obstructive lung diseases.Expert Opin Drug Deliv. 2017 Apr;14(4):483-489. doi: 10.1080/17425247.2016.1223040. Epub 2016 Aug 26. Expert Opin Drug Deliv. 2017. PMID: 27561233 Review.
Cited by
-
Airway Epithelium Dysfunction in Cystic Fibrosis and COPD.Mediators Inflamm. 2018 Apr 8;2018:1309746. doi: 10.1155/2018/1309746. eCollection 2018. Mediators Inflamm. 2018. PMID: 29849481 Free PMC article. Review.
-
In vitro evidence of antioxidant and anti-inflammatory effects of a new nutraceutical formulation explains benefits in a clinical setting of COPD patients.Front Pharmacol. 2024 Aug 20;15:1439835. doi: 10.3389/fphar.2024.1439835. eCollection 2024. Front Pharmacol. 2024. PMID: 39228520 Free PMC article.
-
Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling.Antioxid Redox Signal. 2019 Apr 1;30(10):1331-1351. doi: 10.1089/ars.2017.7403. Epub 2018 Jan 10. Antioxid Redox Signal. 2019. PMID: 29130312 Free PMC article. Review.
-
Cigarette Smoke-Induced Acquired Dysfunction of Cystic Fibrosis Transmembrane Conductance Regulator in the Pathogenesis of Chronic Obstructive Pulmonary Disease.Oxid Med Cell Longev. 2018 Apr 23;2018:6567578. doi: 10.1155/2018/6567578. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 29849907 Free PMC article. Review.
-
Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine.Biomedicines. 2021 Oct 10;9(10):1437. doi: 10.3390/biomedicines9101437. Biomedicines. 2021. PMID: 34680554 Free PMC article. Review.
References
-
- Barnes PJ. New concepts in chronic obstructive pulmonary disease. Annu Rev Med 54: 113–129, 2003 - PubMed
-
- Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 138: 16–27, 2016 - PubMed
-
- Barnes PJ, Shapiro SD, and Pauwels RA. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur Respir J 22: 672–688, 2003 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
