

Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model
- PMID: 28007903
- PMCID: PMC6251521
- DOI: 10.1093/hmg/ddw376
Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model
Abstract
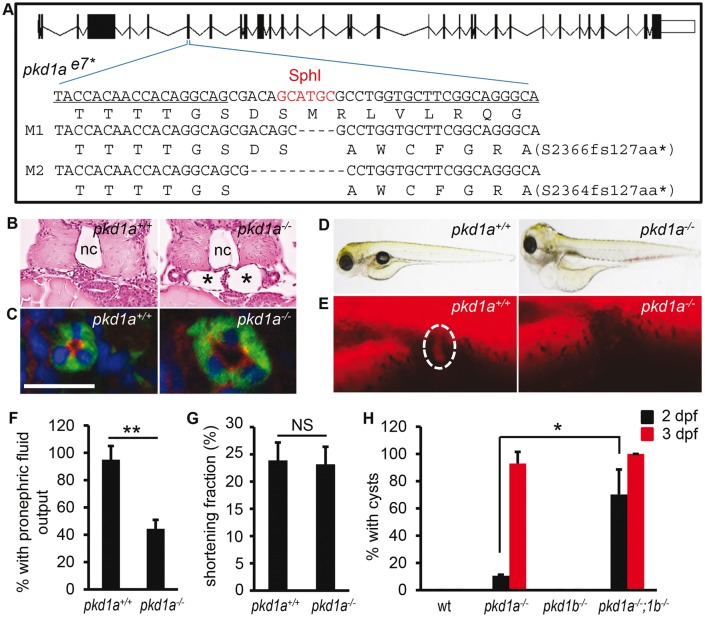
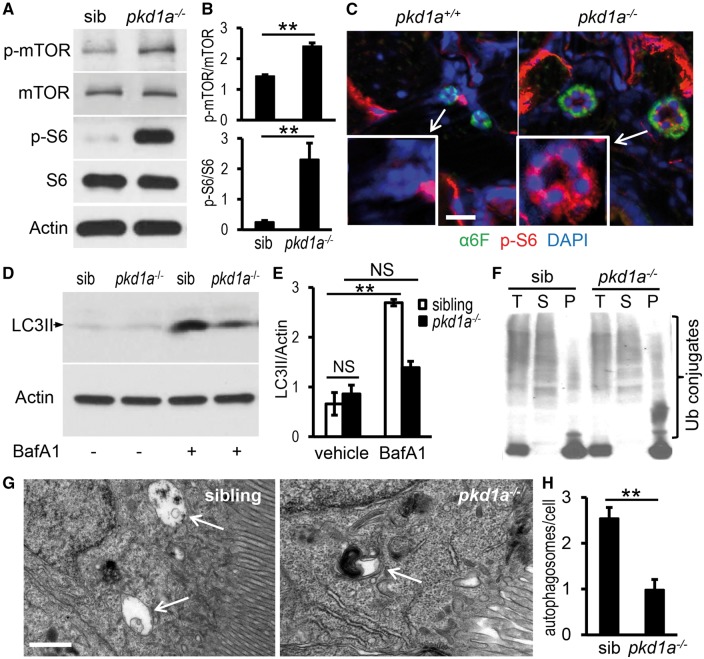
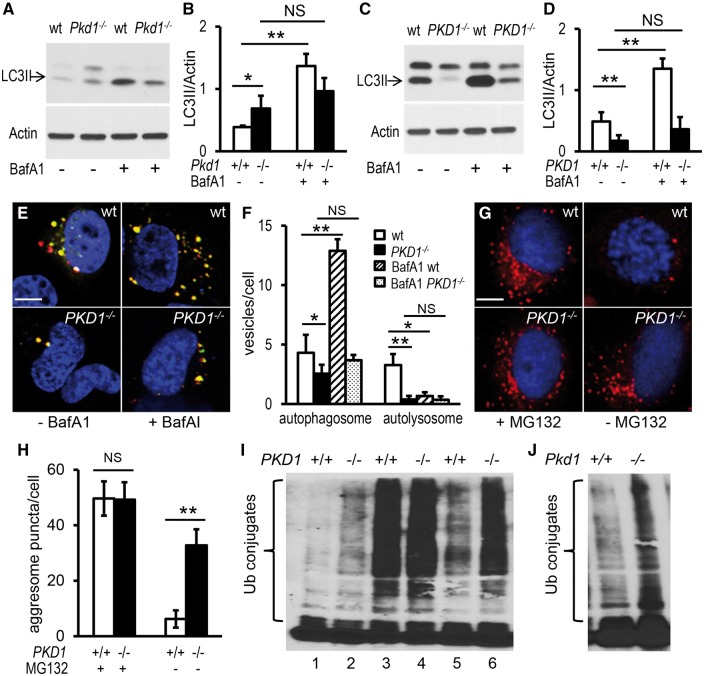
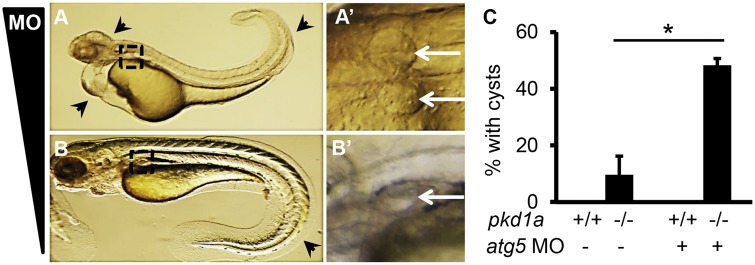
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in either PKD1 or PKD2. It is one of the most common heritable human diseases with eventual development of renal failure; however, effective treatment is lacking. While inhibition of mechanistic target of rapamycin (mTOR) effectively slows cyst expansions in animal models, results from clinical studies are controversial, prompting further mechanistic studies of mTOR-based therapy. Here, we aim to establish autophagy, a downstream pathway of mTOR, as a new therapeutic target for PKD. We generated zebrafish mutants for pkd1 and noted cystic kidney and mTOR activation in pkd1a mutants, suggesting a conserved ADPKD model. Further assessment of the mutants revealed impaired autophagic flux, which was conserved in kidney epithelial cells derived from both Pkd1-null mice and ADPKD patients. We found that inhibition of autophagy by knocking down the core autophagy protein Atg5 promotes cystogenesis, while activation of autophagy using a specific inducer Beclin-1 peptide ameliorates cysts in the pkd1a model. Treatment with compound autophagy activators, including mTOR-dependent rapamycin as well as mTOR-independent carbamazepine and minoxidil, markedly attenuated cyst formation and restored kidney function. Finally, we showed that combination treatment with low doses of rapamycin and carbamazepine was able to attenuate cyst formation as effectively as a single treatment with a high dose of rapamycin alone. In summary, our results suggested a modifying effect of autophagy on ADPKD, established autophagy activation as a novel therapy for ADPKD, and presented zebrafish as an efficient vertebrate model for developing PKD therapeutic strategies.
© The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
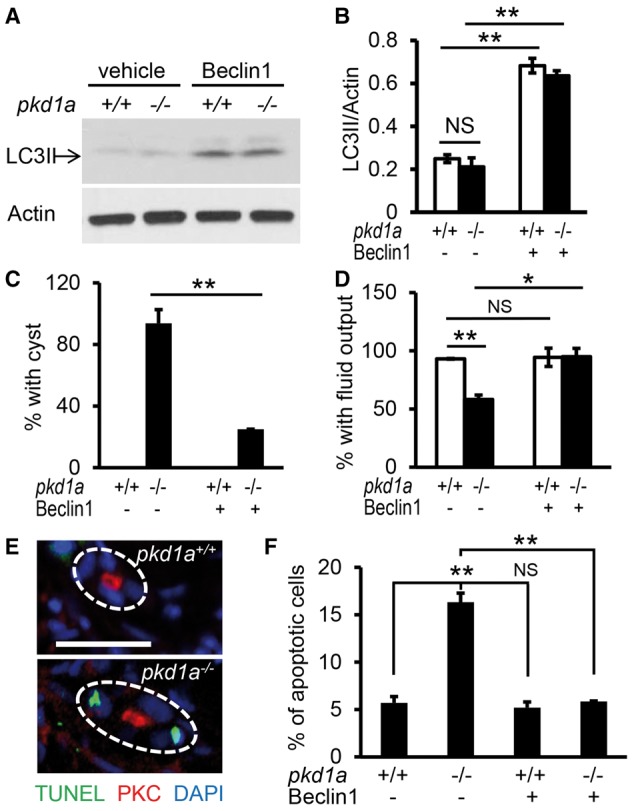
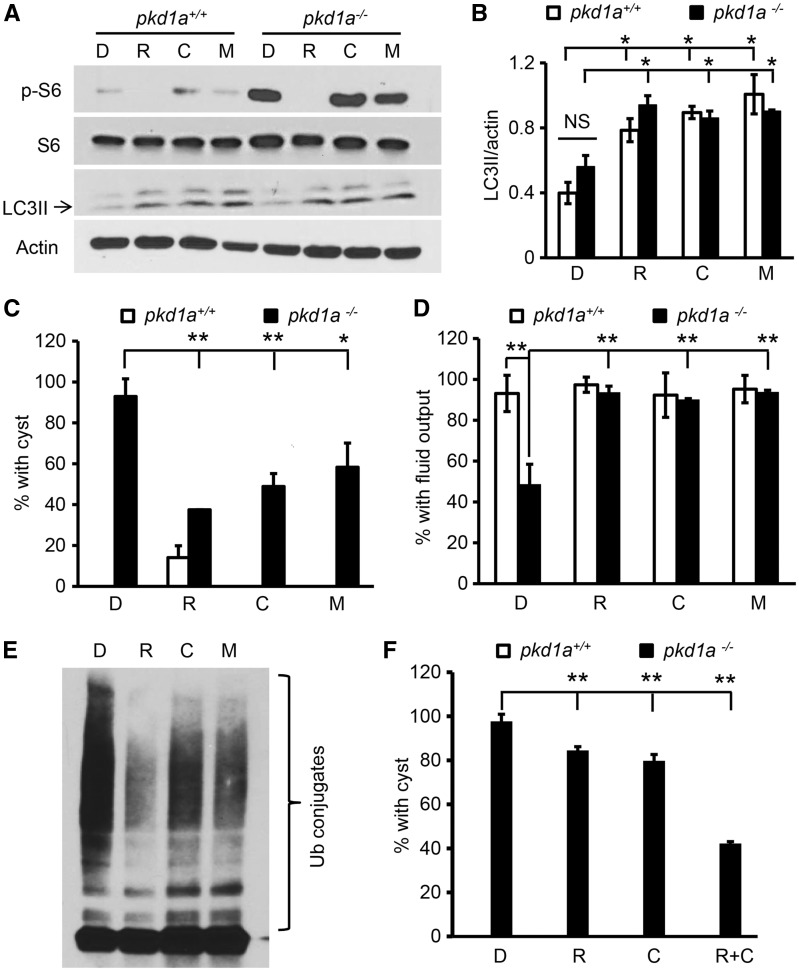
Comment in
-
Polycystic kidney disease: Autophagy boost to treat ADPKD?Nat Rev Nephrol. 2017 Mar;13(3):134. doi: 10.1038/nrneph.2017.1. Epub 2017 Jan 16. Nat Rev Nephrol. 2017. PMID: 28090082 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
