

Cytokine therapy-mediated neuroprotection in a Friedreich's ataxia mouse model
- PMID: 28009062
- PMCID: PMC5324580
- DOI: 10.1002/ana.24846
Cytokine therapy-mediated neuroprotection in a Friedreich's ataxia mouse model
Abstract
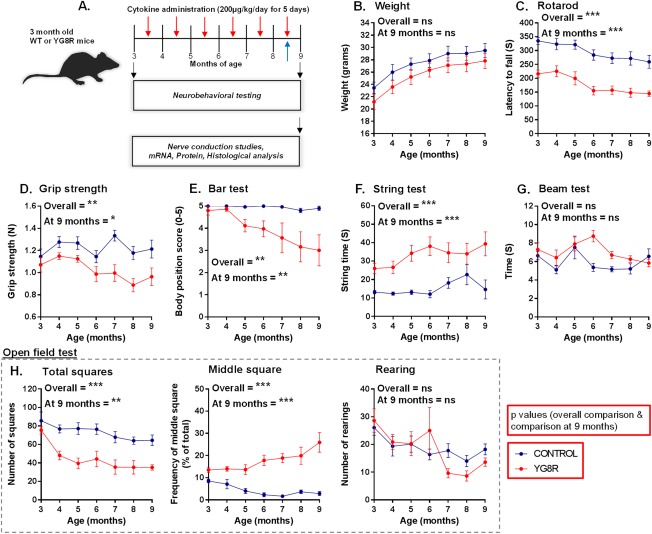
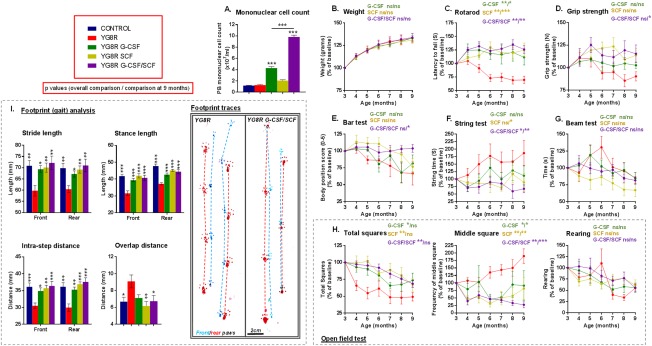
Objectives: Friedreich's ataxia is a devastating neurological disease currently lacking any proven treatment. We studied the neuroprotective effects of the cytokines, granulocyte-colony stimulating factor (G-CSF) and stem cell factor (SCF) in a humanized murine model of Friedreich's ataxia.
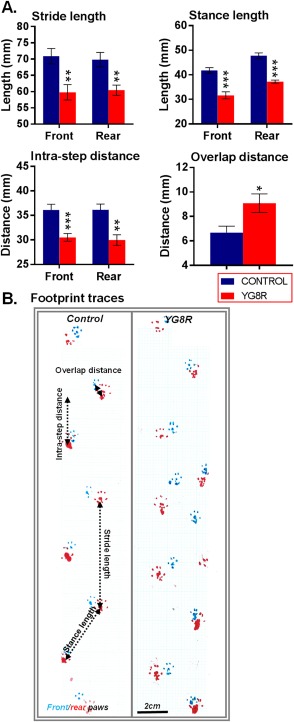
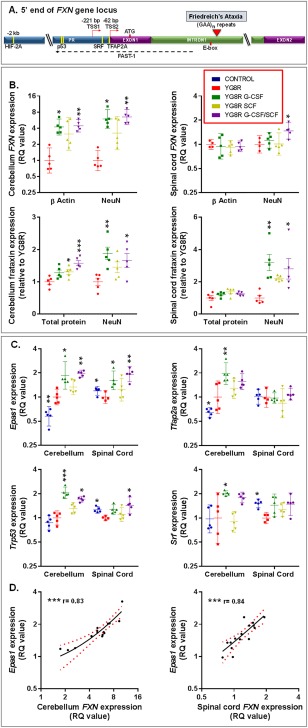
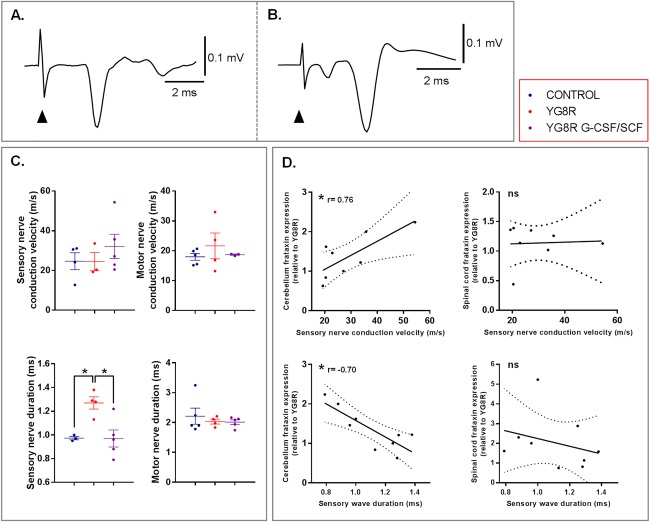
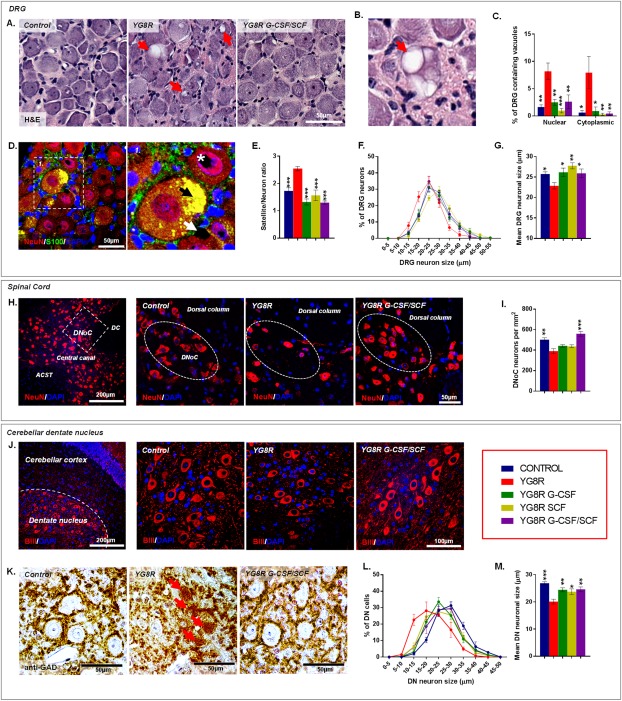
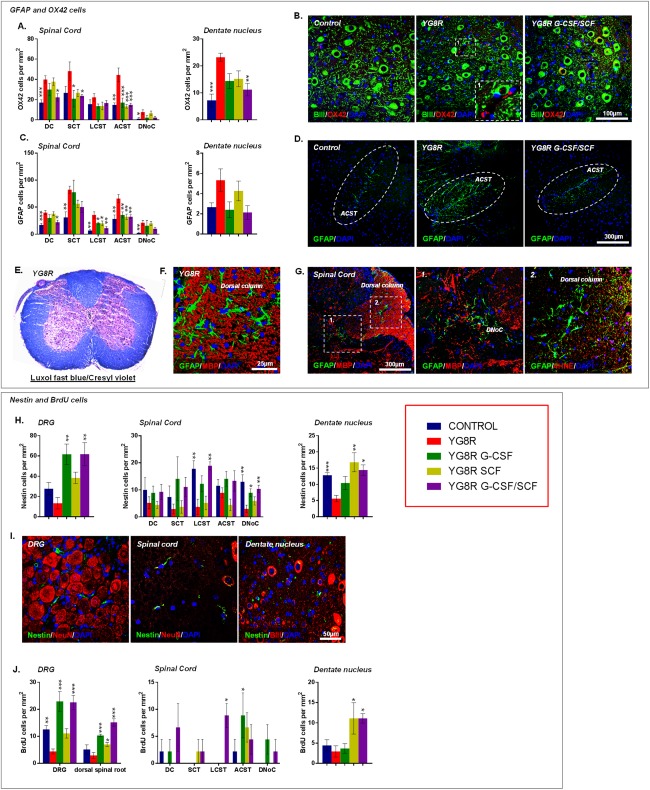
Methods: Mice received monthly subcutaneous infusions of cytokines while also being assessed at monthly time points using an extensive range of behavioral motor performance tests. After 6 months of treatment, neurophysiological evaluation of both sensory and motor nerve conduction was performed. Subsequently, mice were sacrificed for messenger RNA, protein, and histological analysis of the dorsal root ganglia, spinal cord, and cerebellum.
Results: Cytokine administration resulted in significant reversal of biochemical, neuropathological, neurophysiological, and behavioural deficits associated with Friedreich's ataxia. Both G-CSF and SCF had pronounced effects on frataxin levels (the primary molecular defect in the pathogenesis of the disease) and a regulators of frataxin expression. Sustained improvements in motor coordination and locomotor activity were observed, even after onset of neurological symptoms. Treatment also restored the duration of sensory nerve compound potentials. Improvements in peripheral nerve conduction positively correlated with cytokine-induced increases in frataxin expression, providing a link between increases in frataxin and neurophysiological function. Abrogation of disease-related pathology was also evident, with reductions in inflammation/gliosis and increased neural stem cell numbers in areas of tissue injury.
Interpretation: These experiments show that cytokines already clinically used in other conditions offer the prospect of a novel, rapidly translatable, disease-modifying, and neuroprotective treatment for Friedreich's ataxia. Ann Neurol 2017;81:212-226.
© 2017 The Authors. Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Figures
Similar articles
-
Bone marrow transplantation stimulates neural repair in Friedreich's ataxia mice.Ann Neurol. 2018 Apr;83(4):779-793. doi: 10.1002/ana.25207. Ann Neurol. 2018. PMID: 29534309 Free PMC article.
-
Stem cells from wildtype and Friedreich's ataxia mice present similar neuroprotective properties in dorsal root ganglia cells.PLoS One. 2013 May 9;8(5):e62807. doi: 10.1371/journal.pone.0062807. Print 2013. PLoS One. 2013. PMID: 23671637 Free PMC article.
-
Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich's ataxia.Mov Disord. 2019 Mar;34(3):323-334. doi: 10.1002/mds.27604. Epub 2019 Jan 9. Mov Disord. 2019. PMID: 30624801
-
Fixing frataxin: 'ironing out' the metabolic defect in Friedreich's ataxia.Br J Pharmacol. 2014 Apr;171(8):2174-90. doi: 10.1111/bph.12470. Br J Pharmacol. 2014. PMID: 24138602 Free PMC article. Review.
-
Friedreich's ataxia: clinical features, pathogenesis and management.Br Med Bull. 2017 Dec 1;124(1):19-30. doi: 10.1093/bmb/ldx034. Br Med Bull. 2017. PMID: 29053830 Free PMC article. Review.
Cited by
-
Anatomical and functional analysis of the corticospinal tract in an FRDA mouse model.bioRxiv [Preprint]. 2024 Jul 2:2024.06.28.601178. doi: 10.1101/2024.06.28.601178. bioRxiv. 2024. PMID: 39005321 Free PMC article. Preprint.
-
Deep Brain Stimulation of the Interposed Nucleus Reverses Motor Deficits and Stimulates Production of Anti-inflammatory Cytokines in Ataxia Mice.Mol Neurobiol. 2022 Jul;59(7):4578-4592. doi: 10.1007/s12035-022-02872-w. Epub 2022 May 17. Mol Neurobiol. 2022. PMID: 35581519
-
Mesenchymal Stem Cell-Derived Factors Restore Function to Human Frataxin-Deficient Cells.Cerebellum. 2017 Aug;16(4):840-851. doi: 10.1007/s12311-017-0860-y. Cerebellum. 2017. PMID: 28456899 Free PMC article.
-
Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia.Neurotherapeutics. 2019 Oct;16(4):1032-1049. doi: 10.1007/s13311-019-00764-x. Neurotherapeutics. 2019. PMID: 31317428 Free PMC article. Review.
-
Intracerebellar upregulation of Rheb(S16H) ameliorates motor dysfunction in mice with SCA2.Acta Pharmacol Sin. 2025 Jul;46(7):1852-1863. doi: 10.1038/s41401-025-01504-y. Epub 2025 Mar 3. Acta Pharmacol Sin. 2025. PMID: 40033054
References
-
- Harding AE. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981;104:589–620. - PubMed
-
- Campuzano V, Montermini L, Molto MD, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271:1423–1427. - PubMed
-
- Campuzano V, Montermini L, Lutz Y, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 1997;6:1771–1780. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
