

Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations
- PMID: 28009512
- PMCID: PMC5253712
- DOI: 10.1021/jacs.6b11467
Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations
Abstract
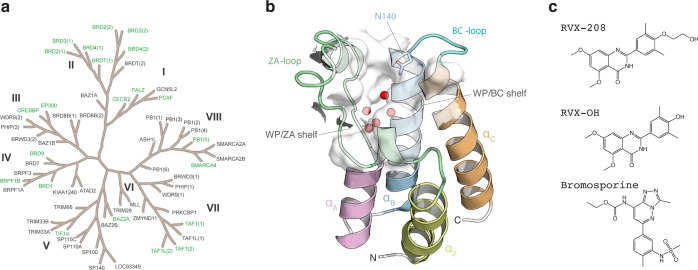
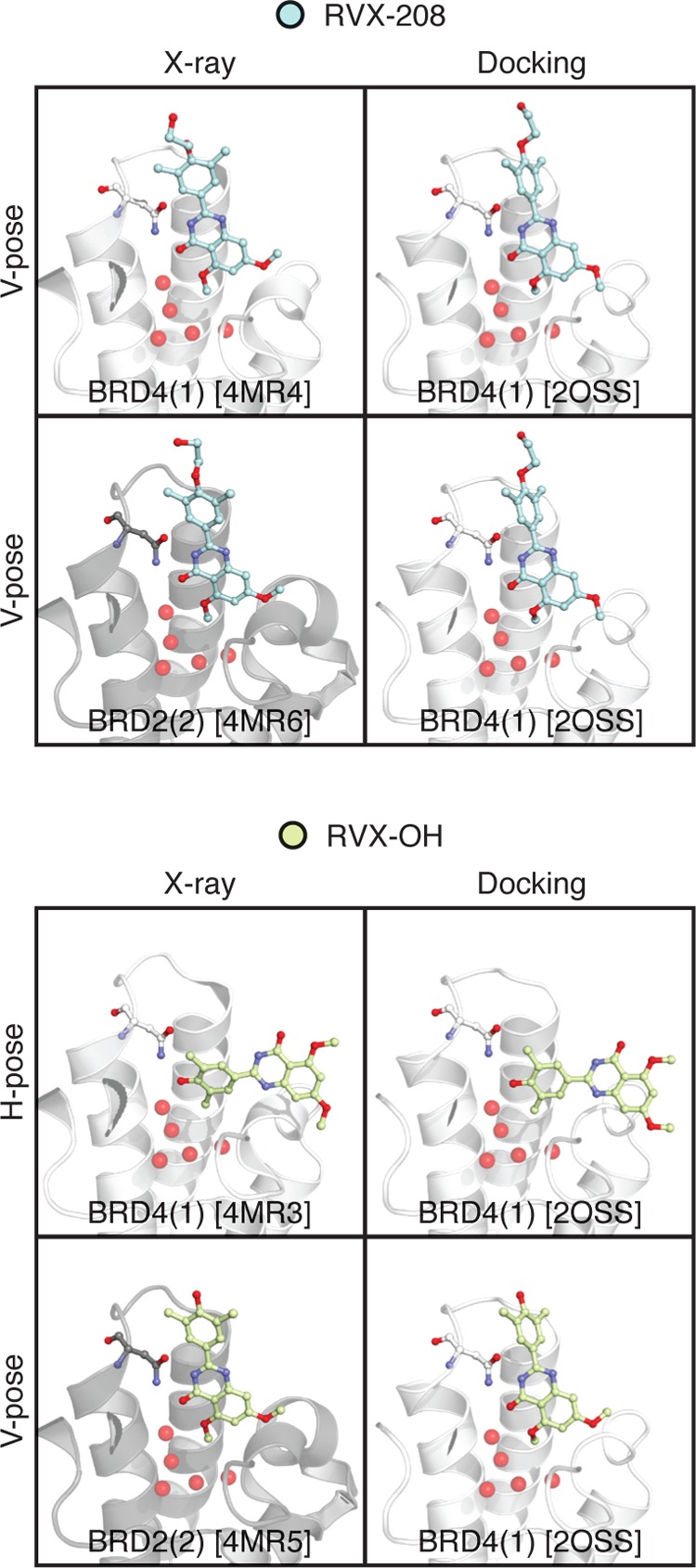
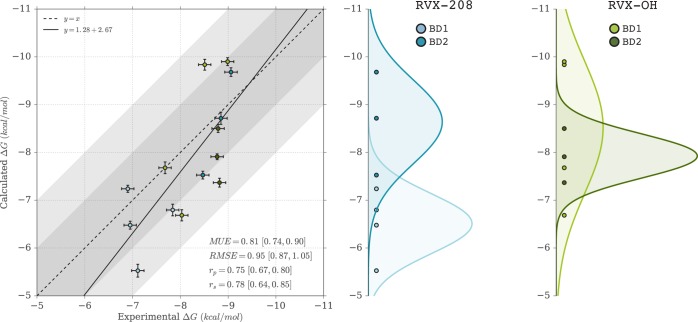
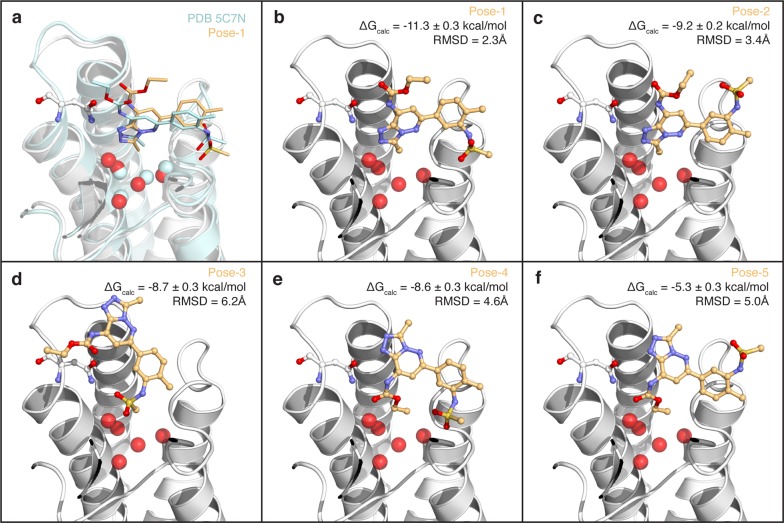
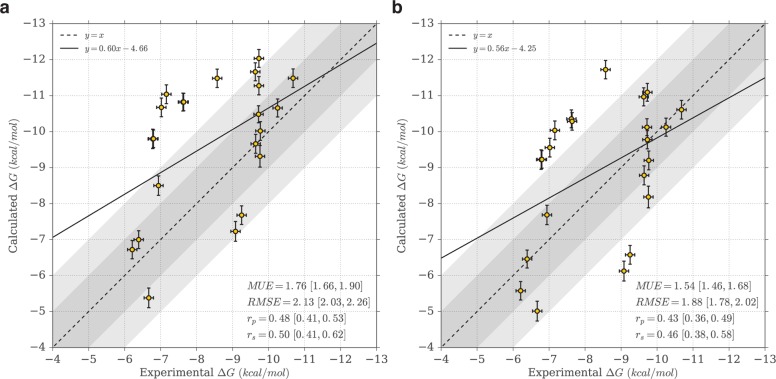
Binding selectivity is a requirement for the development of a safe drug, and it is a critical property for chemical probes used in preclinical target validation. Engineering selectivity adds considerable complexity to the rational design of new drugs, as it involves the optimization of multiple binding affinities. Computationally, the prediction of binding selectivity is a challenge, and generally applicable methodologies are still not available to the computational and medicinal chemistry communities. Absolute binding free energy calculations based on alchemical pathways provide a rigorous framework for affinity predictions and could thus offer a general approach to the problem. We evaluated the performance of free energy calculations based on molecular dynamics for the prediction of selectivity by estimating the affinity profile of three bromodomain inhibitors across multiple bromodomain families, and by comparing the results to isothermal titration calorimetry data. Two case studies were considered. In the first one, the affinities of two similar ligands for seven bromodomains were calculated and returned excellent agreement with experiment (mean unsigned error of 0.81 kcal/mol and Pearson correlation of 0.75). In this test case, we also show how the preferred binding orientation of a ligand for different proteins can be estimated via free energy calculations. In the second case, the affinities of a broad-spectrum inhibitor for 22 bromodomains were calculated and returned a more modest accuracy (mean unsigned error of 1.76 kcal/mol and Pearson correlation of 0.48); however, the reparametrization of a sulfonamide moiety improved the agreement with experiment.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Statistical Analysis on the Performance of Molecular Mechanics Poisson-Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study.J Chem Inf Model. 2017 Sep 25;57(9):2203-2221. doi: 10.1021/acs.jcim.7b00347. Epub 2017 Aug 24. J Chem Inf Model. 2017. PMID: 28786670 Free PMC article.
-
Blind prediction of charged ligand binding affinities in a model binding site.J Mol Biol. 2013 Nov 15;425(22):4569-83. doi: 10.1016/j.jmb.2013.07.030. Epub 2013 Jul 26. J Mol Biol. 2013. PMID: 23896298 Free PMC article.
-
Large scale free energy calculations for blind predictions of protein-ligand binding: the D3R Grand Challenge 2015.J Comput Aided Mol Des. 2016 Sep;30(9):743-751. doi: 10.1007/s10822-016-9952-x. Epub 2016 Aug 25. J Comput Aided Mol Des. 2016. PMID: 27562018 Free PMC article.
-
A Critical Review of Validation, Blind Testing, and Real- World Use of Alchemical Protein-Ligand Binding Free Energy Calculations.Curr Top Med Chem. 2017;17(23):2577-2585. doi: 10.2174/1568026617666170414142131. Curr Top Med Chem. 2017. PMID: 28413950 Review.
-
From bench to bedside, via desktop. Recent advances in the application of cutting-edge in silico tools in the research of drugs targeting bromodomain modules.Biochem Pharmacol. 2019 Jan;159:40-51. doi: 10.1016/j.bcp.2018.11.007. Epub 2018 Nov 9. Biochem Pharmacol. 2019. PMID: 30414936 Review.
Cited by
-
Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs.Proc Natl Acad Sci U S A. 2020 Nov 3;117(44):27381-27387. doi: 10.1073/pnas.2010470117. Epub 2020 Oct 13. Proc Natl Acad Sci U S A. 2020. PMID: 33051297 Free PMC article.
-
An overview of the SAMPL8 host-guest binding challenge.J Comput Aided Mol Des. 2022 Oct;36(10):707-734. doi: 10.1007/s10822-022-00462-5. Epub 2022 Oct 14. J Comput Aided Mol Des. 2022. PMID: 36229622 Free PMC article.
-
Tuning Potential Functions to Host-Guest Binding Data.J Chem Theory Comput. 2024 Jan 9;20(1):239-252. doi: 10.1021/acs.jctc.3c01050. Epub 2023 Dec 26. J Chem Theory Comput. 2024. PMID: 38147689 Free PMC article.
-
Computational screening of FDA approved drugs of fungal origin that may interfere with SARS-CoV-2 spike protein activation, viral RNA replication, and post-translational modification: a multiple target approach.In Silico Pharmacol. 2021 Apr 4;9(1):27. doi: 10.1007/s40203-021-00089-8. eCollection 2021. In Silico Pharmacol. 2021. PMID: 33842191 Free PMC article.
-
AMBER Free Energy Tools: A New Framework for the Design of Optimized Alchemical Transformation Pathways.J Chem Theory Comput. 2023 Jan 9:10.1021/acs.jctc.2c00725. doi: 10.1021/acs.jctc.2c00725. Online ahead of print. J Chem Theory Comput. 2023. PMID: 36622640 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
