

Outside in: The matrix as a modifier of muscular dystrophy
- PMID: 28011285
- PMCID: PMC5262521
- DOI: 10.1016/j.bbamcr.2016.12.020
Outside in: The matrix as a modifier of muscular dystrophy
Abstract
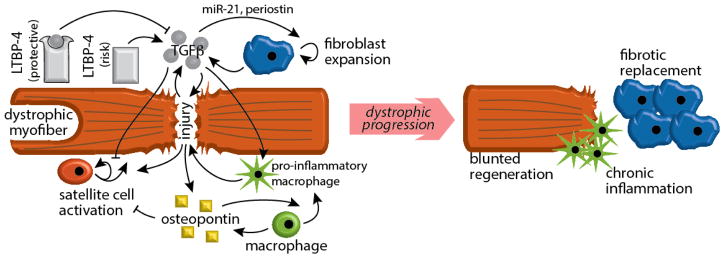
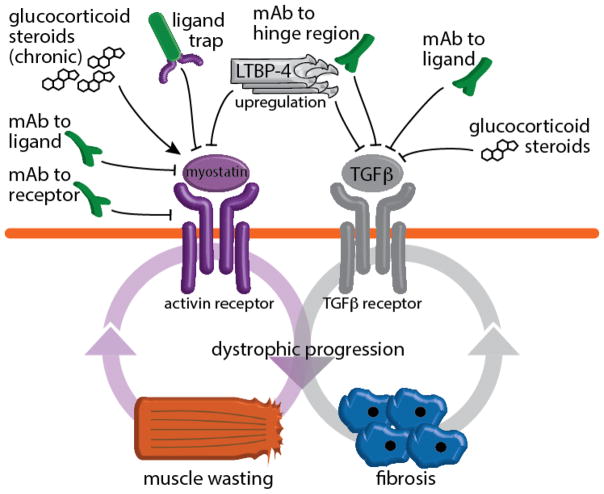
Muscular dystrophies are genetic conditions leading to muscle degeneration and often, impaired regeneration. Duchenne Muscular Dystrophy is a prototypical form of muscular dystrophy, and like other forms of genetically inherited muscle diseases, pathological progression is variable. Variability in muscular dystrophy can arise from differences in the manner in which the primary mutation impacts the affected protein's function; however, clinical heterogeneity also derives from secondary mutations in other genes that can enhance or reduce pathogenic features of disease. These genes, called genetic modifiers, regulate the pathophysiological context of dystrophic degeneration and regeneration. Understanding the mechanistic links between genetic modifiers and dystrophic progression sheds light on pathologic remodeling, and provides novel avenues to therapeutically intervene to reduce muscle degeneration. Based on targeted genetic approaches and unbiased genomewide screens, several modifiers have been identified for muscular dystrophy, including extracellular agonists of signaling cascades. This review will focus on identification and possible mechanisms of recently identified modifiers for muscular dystrophy, including osteopontin, latent TGFβ binding protein 4 (LTBP4) and Jagged1. Moreover, we will review the investigational approaches that aim to target modifier pathways and thereby counteract dystrophic muscle wasting.
Keywords: Duchenne Muscular Dystrophy; Genetic modifiers; Investigational medicinal products; Jagged1; LTBP4; Monoclonal antibodies; Myostatin; Notch; Novel drugs; Osteopontin; SPP1; TGFβ.
Copyright © 2016 Elsevier B.V. All rights reserved.
Conflict of interest statement
EMM has provided consulting services for Novartis, Invitae, Mitobridge, Summitplc, AstraZeneca, and Pfizer and served as a data safety monitor for Eli Lilly and Fibrogen, and has patent application 13/957,100 “Mitigating tissue damage and fibrosis via latent TGFβ protein (LTBP4).
Figures
Similar articles
-
Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics.Matrix Biol. 2018 Aug;68-69:616-627. doi: 10.1016/j.matbio.2018.02.016. Epub 2018 Feb 23. Matrix Biol. 2018. PMID: 29481844 Free PMC article. Review.
-
Modifier genes and their effect on Duchenne muscular dystrophy.Curr Opin Neurol. 2015 Oct;28(5):528-34. doi: 10.1097/WCO.0000000000000240. Curr Opin Neurol. 2015. PMID: 26263473 Free PMC article. Review.
-
Overexpression of Latent TGFβ Binding Protein 4 in Muscle Ameliorates Muscular Dystrophy through Myostatin and TGFβ.PLoS Genet. 2016 May 5;12(5):e1006019. doi: 10.1371/journal.pgen.1006019. eCollection 2016 May. PLoS Genet. 2016. PMID: 27148972 Free PMC article.
-
Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury.PLoS Genet. 2017 Oct 24;13(10):e1007070. doi: 10.1371/journal.pgen.1007070. eCollection 2017 Oct. PLoS Genet. 2017. PMID: 29065150 Free PMC article.
-
Anti-latent TGFβ binding protein 4 antibody improves muscle function and reduces muscle fibrosis in muscular dystrophy.Sci Transl Med. 2021 Sep 8;13(610):eabf0376. doi: 10.1126/scitranslmed.abf0376. Epub 2021 Sep 8. Sci Transl Med. 2021. PMID: 34516828 Free PMC article.
Cited by
-
Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone.Skelet Muscle. 2018 Oct 27;8(1):34. doi: 10.1186/s13395-018-0180-z. Skelet Muscle. 2018. PMID: 30368252 Free PMC article.
-
Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics.Matrix Biol. 2018 Aug;68-69:616-627. doi: 10.1016/j.matbio.2018.02.016. Epub 2018 Feb 23. Matrix Biol. 2018. PMID: 29481844 Free PMC article. Review.
-
Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification.Toxicol Pathol. 2017 Oct;45(7):961-976. doi: 10.1177/0192623317734823. Epub 2017 Oct 3. Toxicol Pathol. 2017. PMID: 28974147 Free PMC article. Review.
-
Bacteroides plebeius improves muscle wasting in chronic kidney disease by modulating the gut-renal muscle axis.J Cell Mol Med. 2022 Dec;26(24):6066-6078. doi: 10.1111/jcmm.17626. Epub 2022 Dec 2. J Cell Mol Med. 2022. PMID: 36458537 Free PMC article.
-
Genetic manipulation of CCN2/CTGF unveils cell-specific ECM-remodeling effects in injured skeletal muscle.FASEB J. 2019 Feb;33(2):2047-2057. doi: 10.1096/fj.201800622RR. Epub 2018 Sep 14. FASEB J. 2019. PMID: 30216109 Free PMC article.
References
-
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. - PubMed
-
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. - PubMed
-
- Mercier S, Toutain A, Toussaint A, Raynaud M, de Barace C, Marcorelles P, Pasquier L, Blayau M, Espil C, Parent P, Journel H, Lazaro L, Andoni Urtizberea J, Moerman A, Faivre L, Eymard B, Maincent K, Gherardi R, Chaigne D, Ben Yaou R, Leturcq F, Chelly J, Desguerre I. Genetic and clinical specificity of 26 symptomatic carriers for dystrophinopathies at pediatric age. Eur J Hum Genet. 2013;21:855–863. - PMC - PubMed
-
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, Weiss RB C. United Dystrophinopathy Project. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
