

Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome
- PMID: 28013382
- PMCID: PMC5400701
- DOI: 10.1007/s00467-016-3565-4
Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome
Abstract
Background: Many COL4A5 splice region variants have been described in patients with X-linked Alport syndrome, but few have been confirmed by functional analysis to actually cause defective splicing. We sought to demonstrate that a novel COL4A5 splice region variant in a family with Alport syndrome is pathogenic using functional studies. We also describe an alternative method of diagnosis.
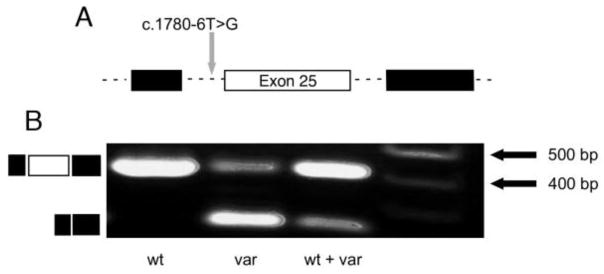
Methods: Targeted next-generation sequencing results of an individual with Alport syndrome were analyzed and the results confirmed by Sanger sequencing in family members. A splicing reporter minigene assay was used to examine the variant's effect on splicing in transfected cells. Plucked hair follicles from patients and controls were examined for collagen IV proteins using immunofluorescence microscopy.
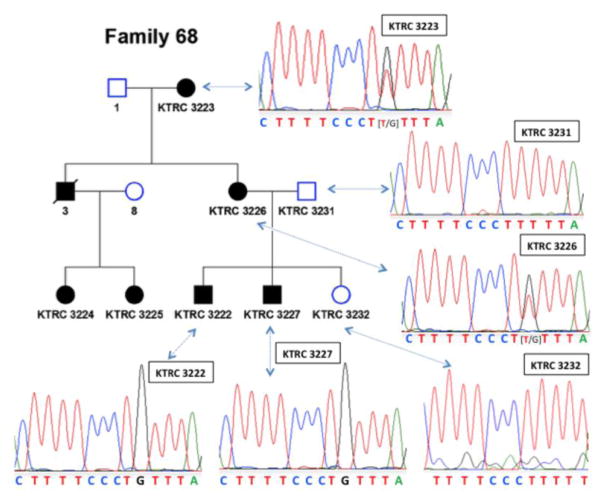
Results: A novel splice region mutation in COL4A5, c.1780-6T>G, was identified and segregated with disease in this family. This variant caused frequent skipping of exon 25, resulting in a frameshift and truncation of collagen α5(IV) protein. We also developed and validated a new approach to characterize the expression of collagen α5(IV) protein in the basement membranes of plucked hair follicles. Using this approach we demonstrated reduced collagen α5(IV) protein in affected male and female individuals in this family, supporting frequent failure of normal splicing.
Conclusions: Differing normal to abnormal transcript ratios in affected individuals carrying splice region variants may contribute to variable disease severity observed in Alport families. Examination of plucked hair follicles in suspected X-linked Alport syndrome patients may offer a less invasive alternative method of diagnosis and serve as a pathogenicity test for COL4A5 variants of uncertain significance.
Keywords: Alport syndrome; COL4A5; Hair follicle; Immunofluorescence; Splice site mutation.
Figures

References
-
- Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore) 1999;78:338–360. - PubMed
-
- Tryggvason K, Zhou J, Hostikka SL, Shows TB. Molecular genetics of Alport syndrome. Kidney Int. 1993;43:38–44. - PubMed
-
- Renieri A, Meroni M, Sessa A, Battini G, Serbelloni P, Torri Tarelli L, Seri M, Galli L, De Marchi M. Variability of clinical phenotype in a large Alport family with Gly 1143 Ser change of collagen alpha 5(IV)-chain. Nephron. 1994;67:444–449. - PubMed
-
- Tsiakkis D, Pieri M, Koupepidou P, Demosthenous P, Panayidou K, Deltas C. Genotype-phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin Genet. 2012;82:297–299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
