

Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers
- PMID: 28017794
- PMCID: PMC5500296
- DOI: 10.1016/j.stem.2016.08.019
Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers
Abstract
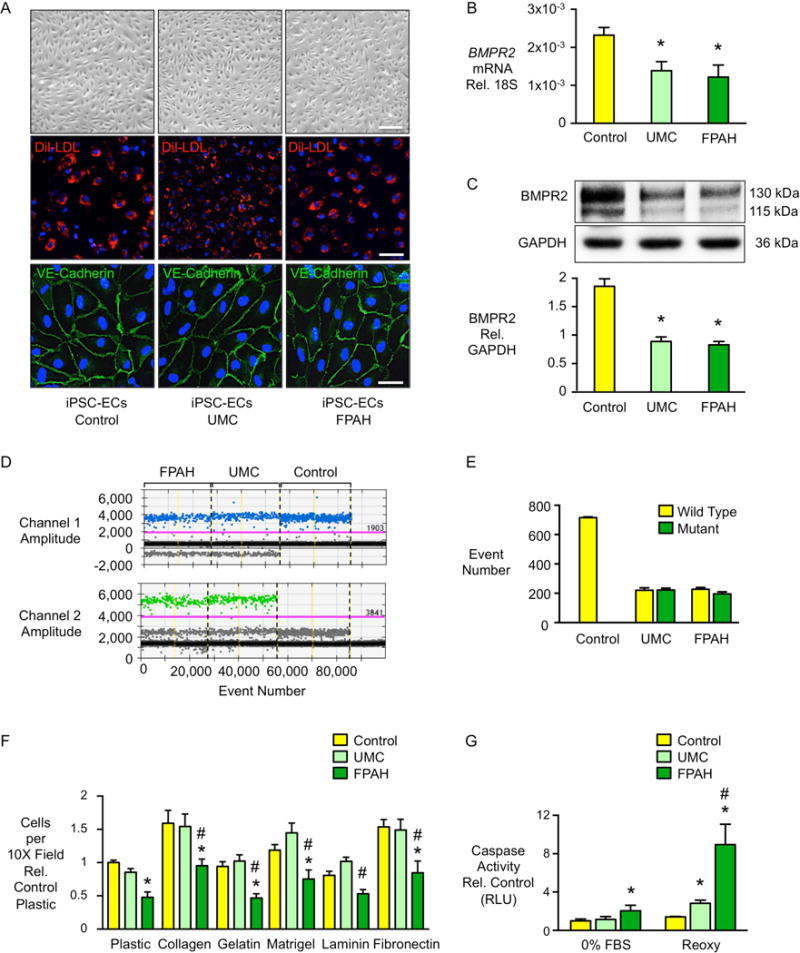
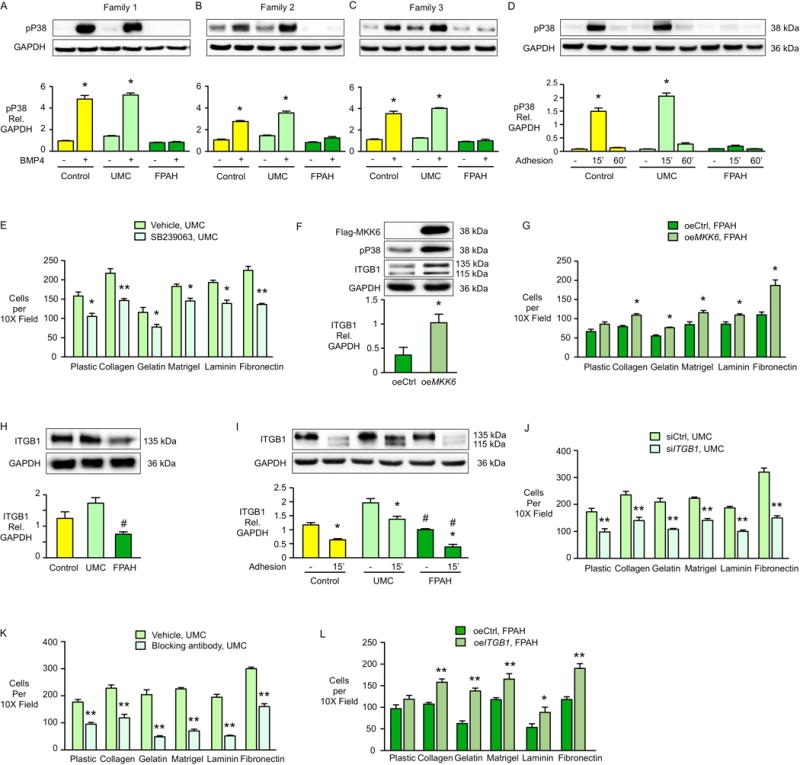
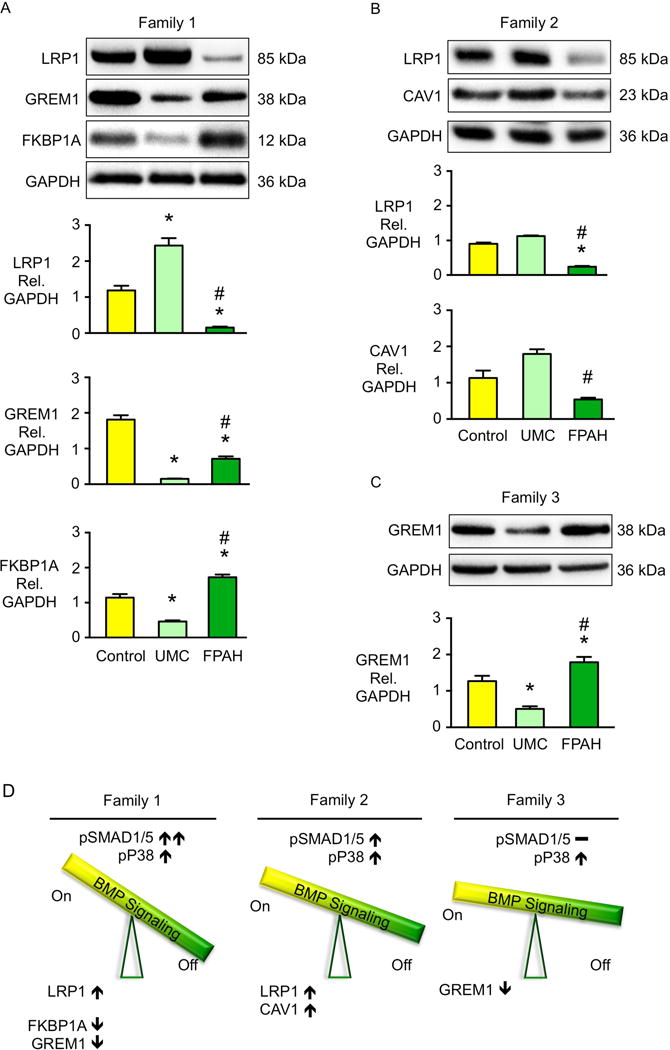
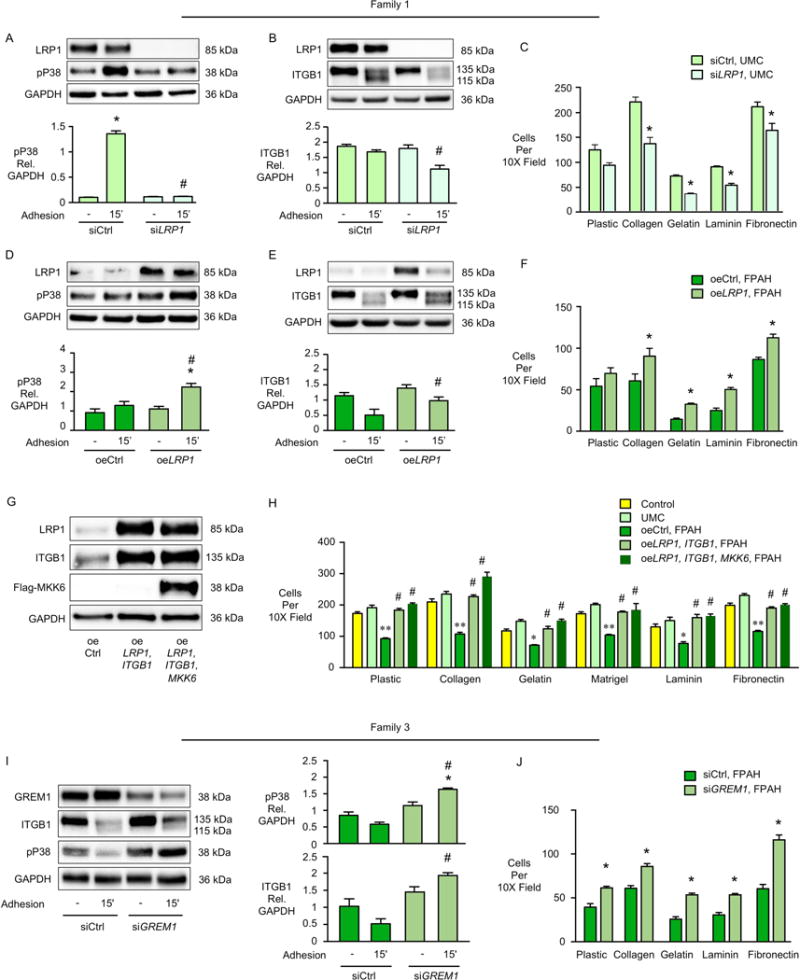
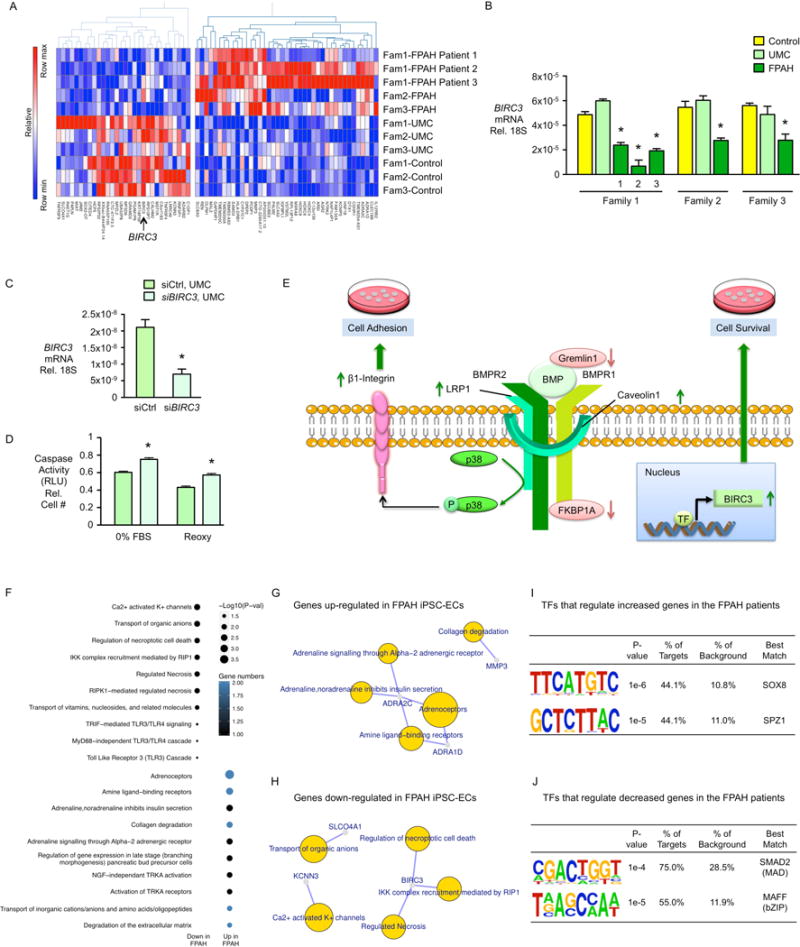
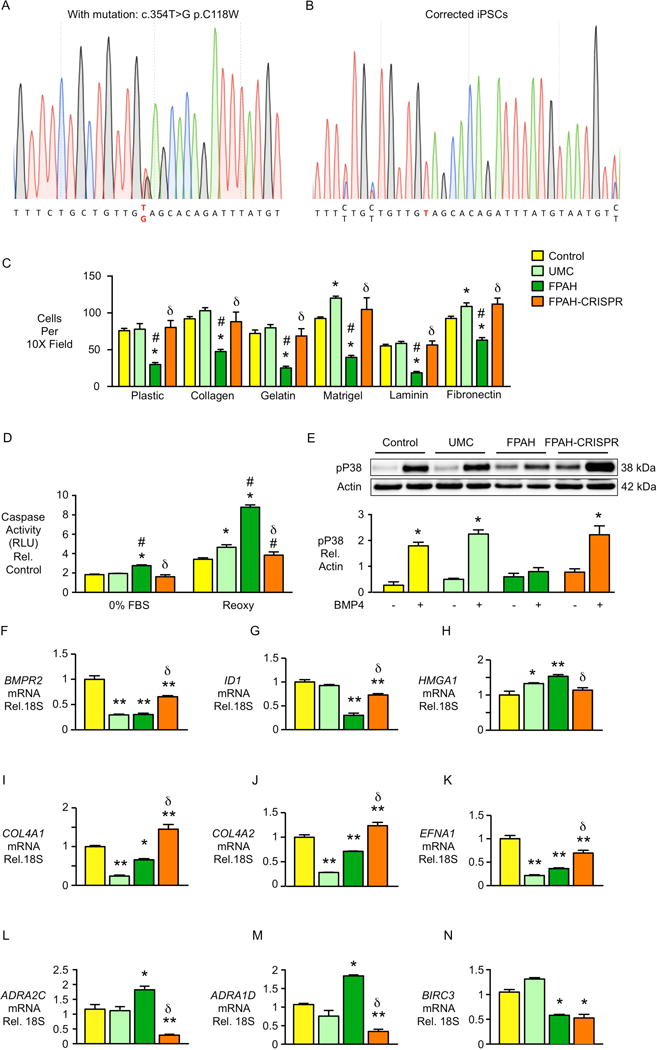
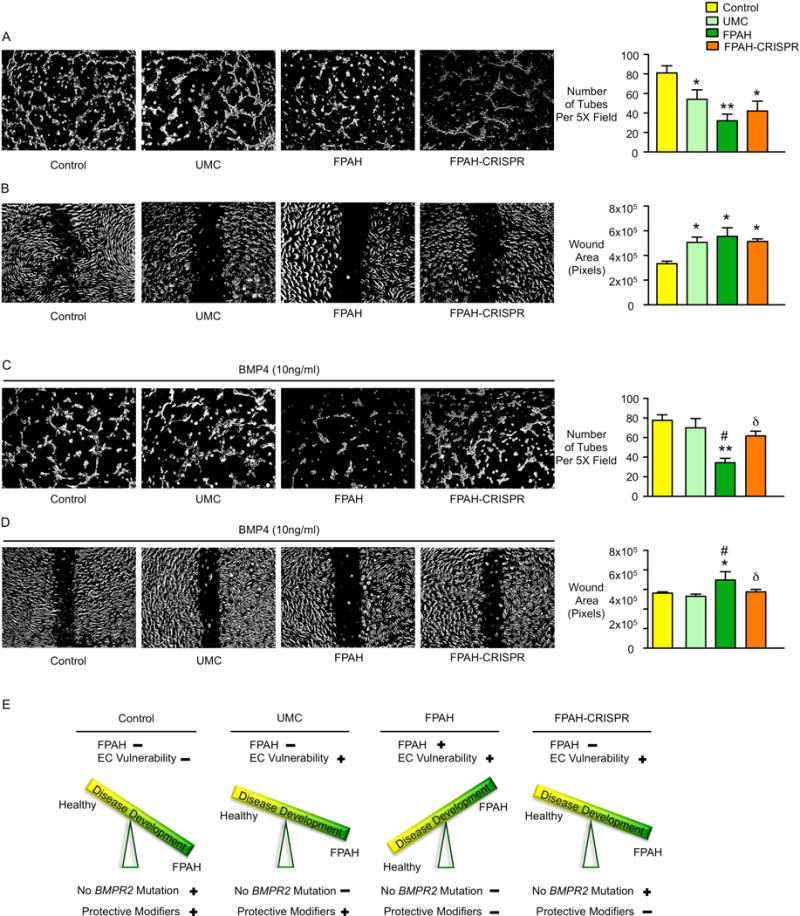
In familial pulmonary arterial hypertension (FPAH), the autosomal dominant disease-causing BMPR2 mutation is only 20% penetrant, suggesting that genetic variation provides modifiers that alleviate the disease. Here, we used comparison of induced pluripotent stem cell-derived endothelial cells (iPSC-ECs) from three families with unaffected mutation carriers (UMCs), FPAH patients, and gender-matched controls to investigate this variation. Our analysis identified features of UMC iPSC-ECs related to modifiers of BMPR2 signaling or to differentially expressed genes. FPAH-iPSC-ECs showed reduced adhesion, survival, migration, and angiogenesis compared to UMC-iPSC-ECs and control cells. The "rescued" phenotype of UMC cells was related to an increase in specific BMPR2 activators and/or a reduction in inhibitors, and the improved cell adhesion could be attributed to preservation of related signaling. The improved survival was related to increased BIRC3 and was independent of BMPR2. Our findings therefore highlight protective modifiers for FPAH that could help inform development of future treatment strategies.
Keywords: bone morphogenetic protein receptor 2; cell adhesion; cell signaling; cell survival; endothelial dysfunction; induced pluripotent stem cell-derived endothelial cell; penetrance; pulmonary arterial hypertension; transcriptomic analysis; unaffected mutation carrier.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have no conflicting financial interests.
Figures
Comment in
-
Putting skin in the game: dermis-derived stem cells provide insight into familial pulmonary hypertension.Stem Cell Investig. 2017 May 5;4:35. doi: 10.21037/sci.2017.04.05. eCollection 2017. Stem Cell Investig. 2017. PMID: 28607909 Free PMC article. No abstract available.
-
Inducible pluripotent stem cells and pulmonary arterial hypertension: the future is now!Stem Cell Investig. 2017 Jun 13;4:53. doi: 10.21037/sci.2017.05.10. eCollection 2017. Stem Cell Investig. 2017. PMID: 28725649 Free PMC article. No abstract available.
-
Rescuing BMPR2-driven endothelial dysfunction in PAH: a novel treatment strategy for the future?Stem Cell Investig. 2017 Jun 14;4:56. doi: 10.21037/sci.2017.05.11. eCollection 2017. Stem Cell Investig. 2017. PMID: 28725652 Free PMC article. No abstract available.
References
-
- Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. - PubMed
-
- Cahill E, Costello CM, Rowan SC, Harkin S, Howell K, Leonard MO, Southwood M, Cummins EP, Fitzpatrick SF, Taylor CT, et al. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation. 2012;125:920–930. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
