

Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity
- PMID: 28017796
- PMCID: PMC5384872
- DOI: 10.1016/j.stem.2016.11.005
Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity
Erratum in
-
Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity.Cell Stem Cell. 2022 Oct 6;29(10):1505. doi: 10.1016/j.stem.2022.08.011. Cell Stem Cell. 2022. PMID: 36206733 No abstract available.
Abstract
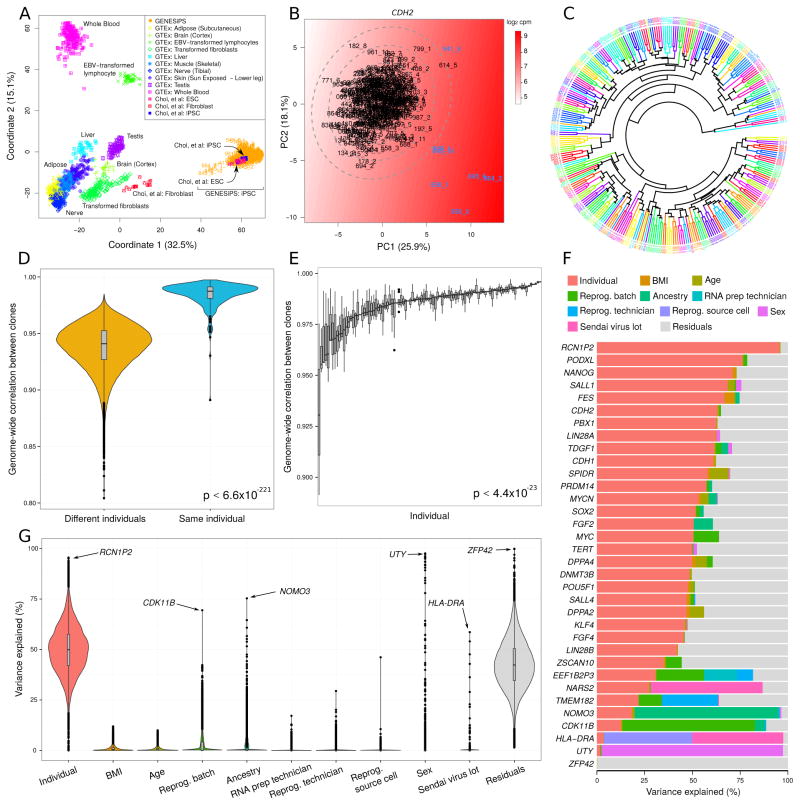
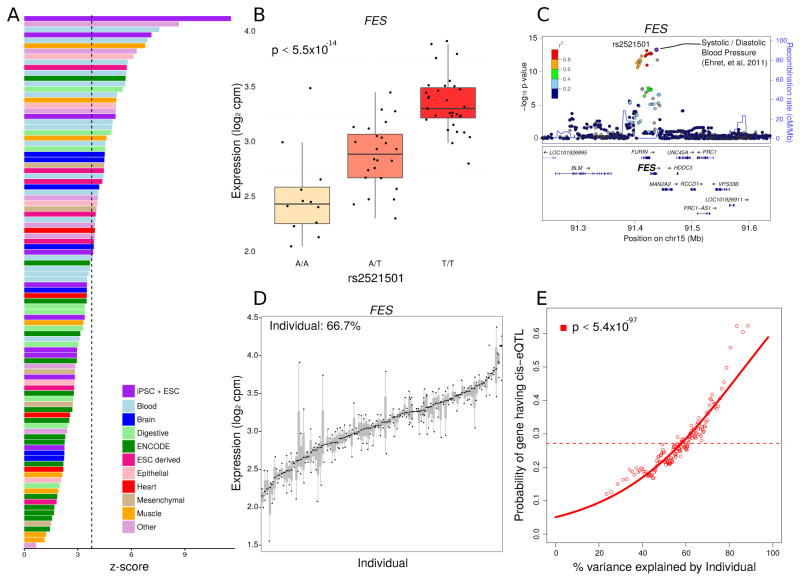
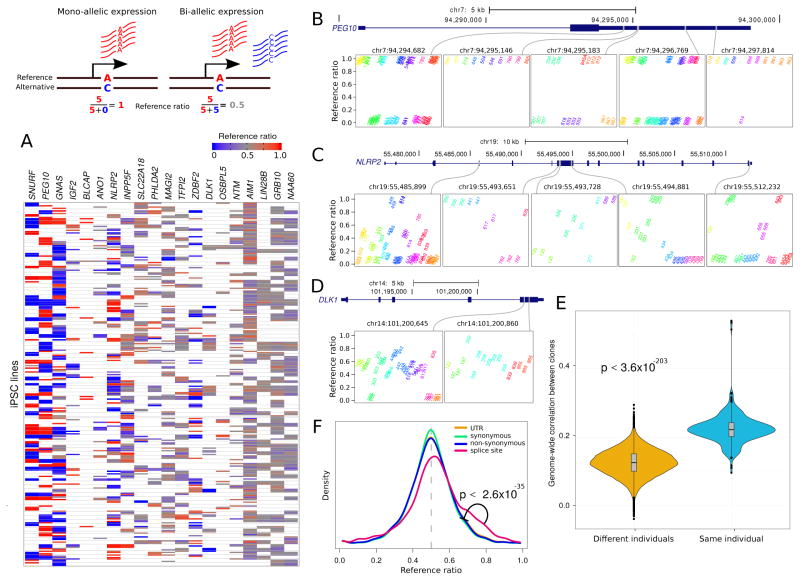
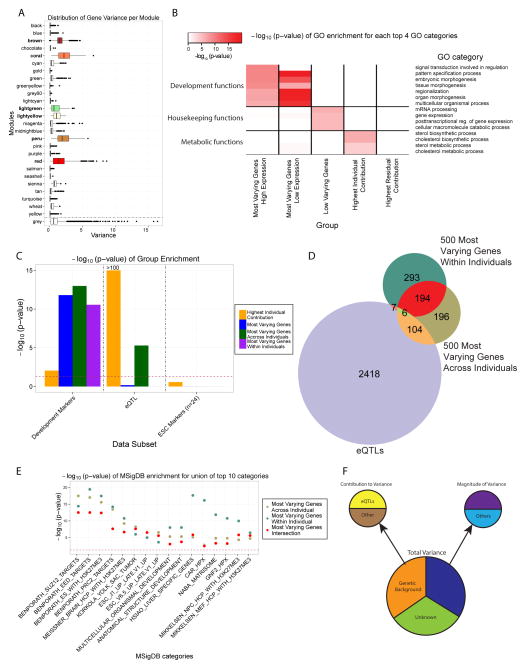
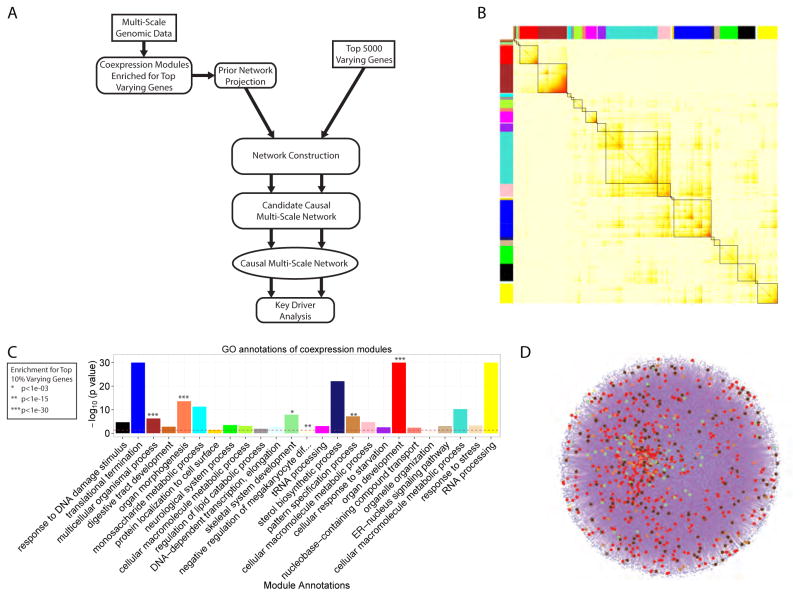
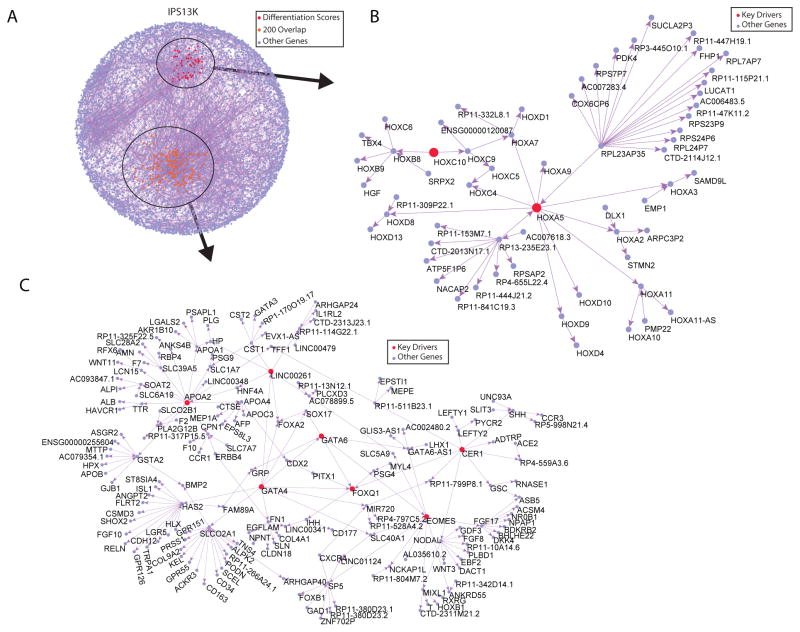
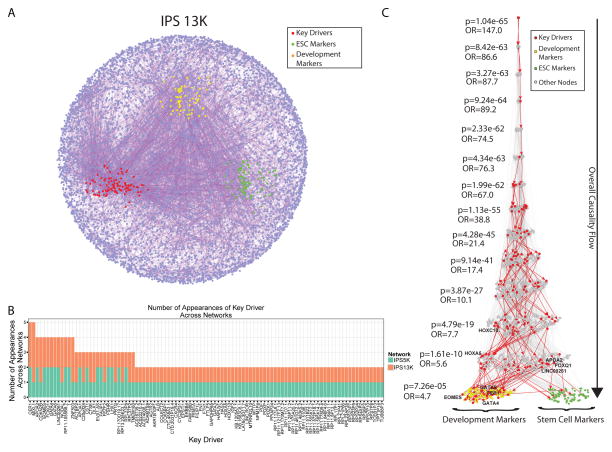
Variability in induced pluripotent stem cell (iPSC) lines remains a concern for disease modeling and regenerative medicine. We have used RNA-sequencing analysis and linear mixed models to examine the sources of gene expression variability in 317 human iPSC lines from 101 individuals. We found that ∼50% of genome-wide expression variability is explained by variation across individuals and identified a set of expression quantitative trait loci that contribute to this variation. These analyses coupled with allele-specific expression show that iPSCs retain a donor-specific gene expression pattern. Network, pathway, and key driver analyses showed that Polycomb targets contribute significantly to the non-genetic variability seen within and across individuals, highlighting this chromatin regulator as a likely source of reprogramming-based variability. Our findings therefore shed light on variation between iPSC lines and illustrate the potential for our dataset and other similar large-scale analyses to identify underlying drivers relevant to iPSC applications.
Keywords: Polycomb targets; allelic imbalance; differentiation variability; eQTL; iPSC library; key drivers; network analysis; transcriptional variability; variance partition.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. 2013;140:2525–2534. - PubMed
-
- Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell stem cell. 2011;9:17–23. - PubMed
-
- Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell stem cell. 2011;9:97–102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
