

Stress Signals, Mediated by Membranous Glucocorticoid Receptor, Activate PLC/PKC/GSK-3β/β-catenin Pathway to Inhibit Wound Closure
- PMID: 28017831
- PMCID: PMC7540219
- DOI: 10.1016/j.jid.2016.11.036
Stress Signals, Mediated by Membranous Glucocorticoid Receptor, Activate PLC/PKC/GSK-3β/β-catenin Pathway to Inhibit Wound Closure
Abstract
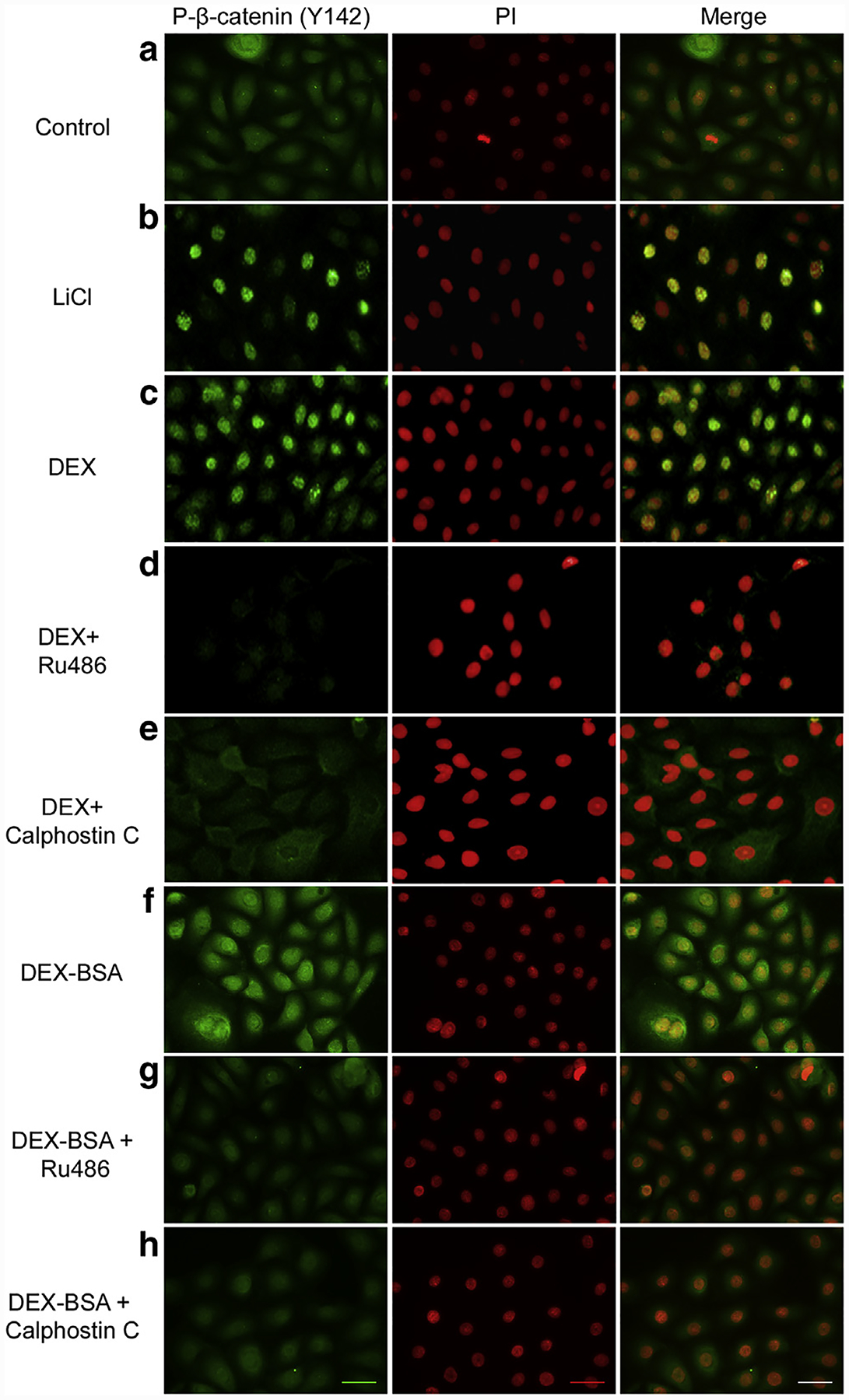
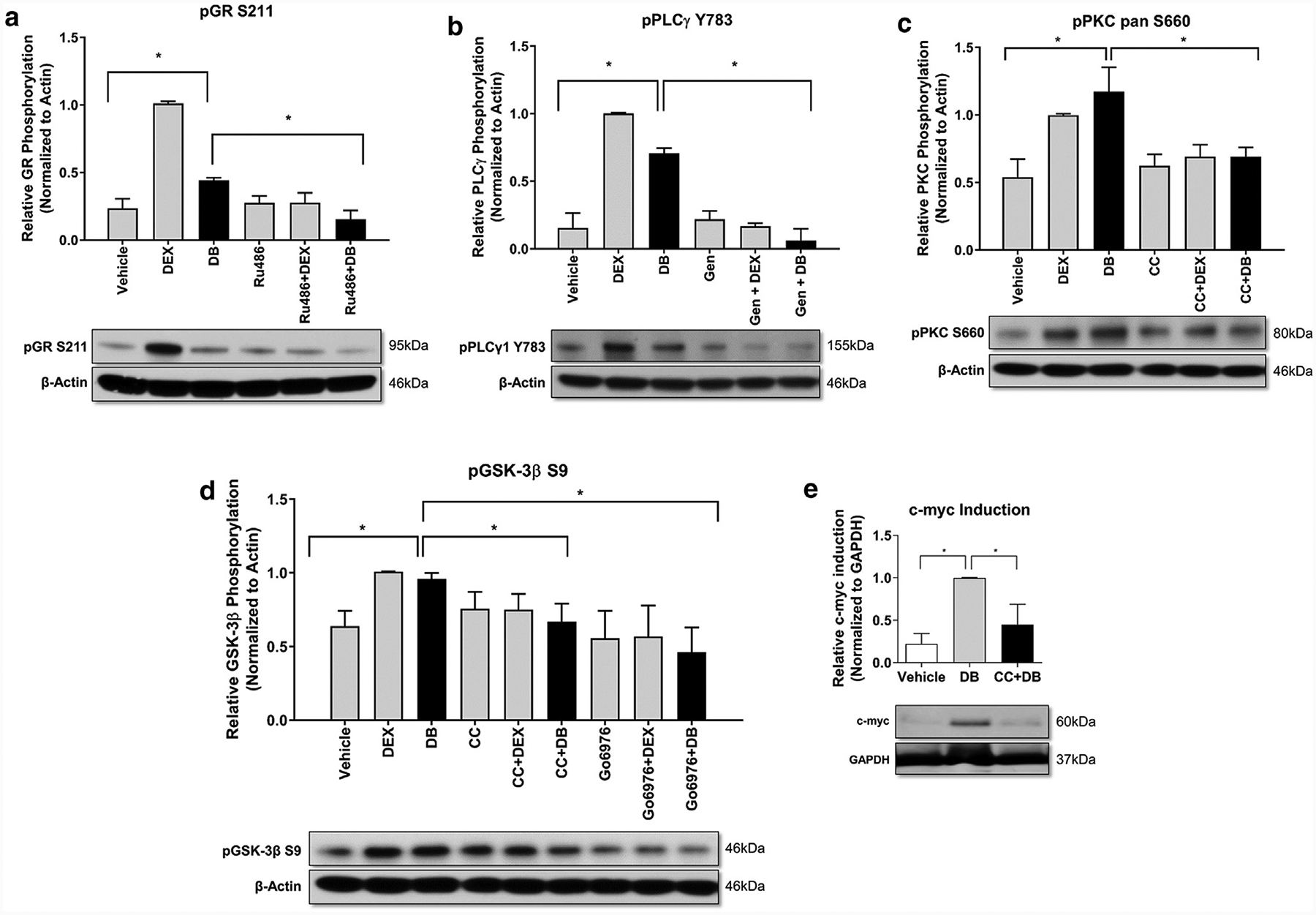
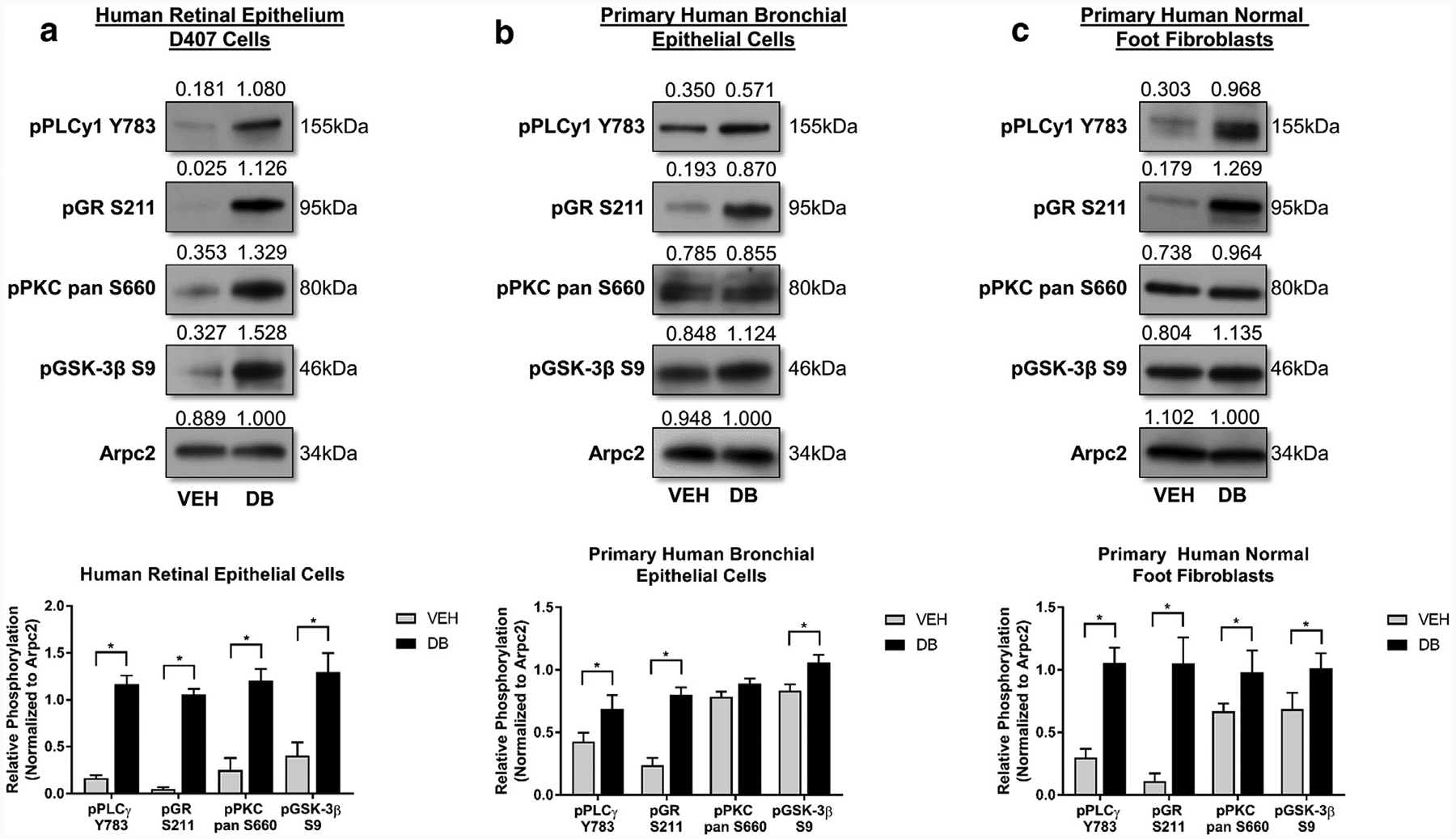
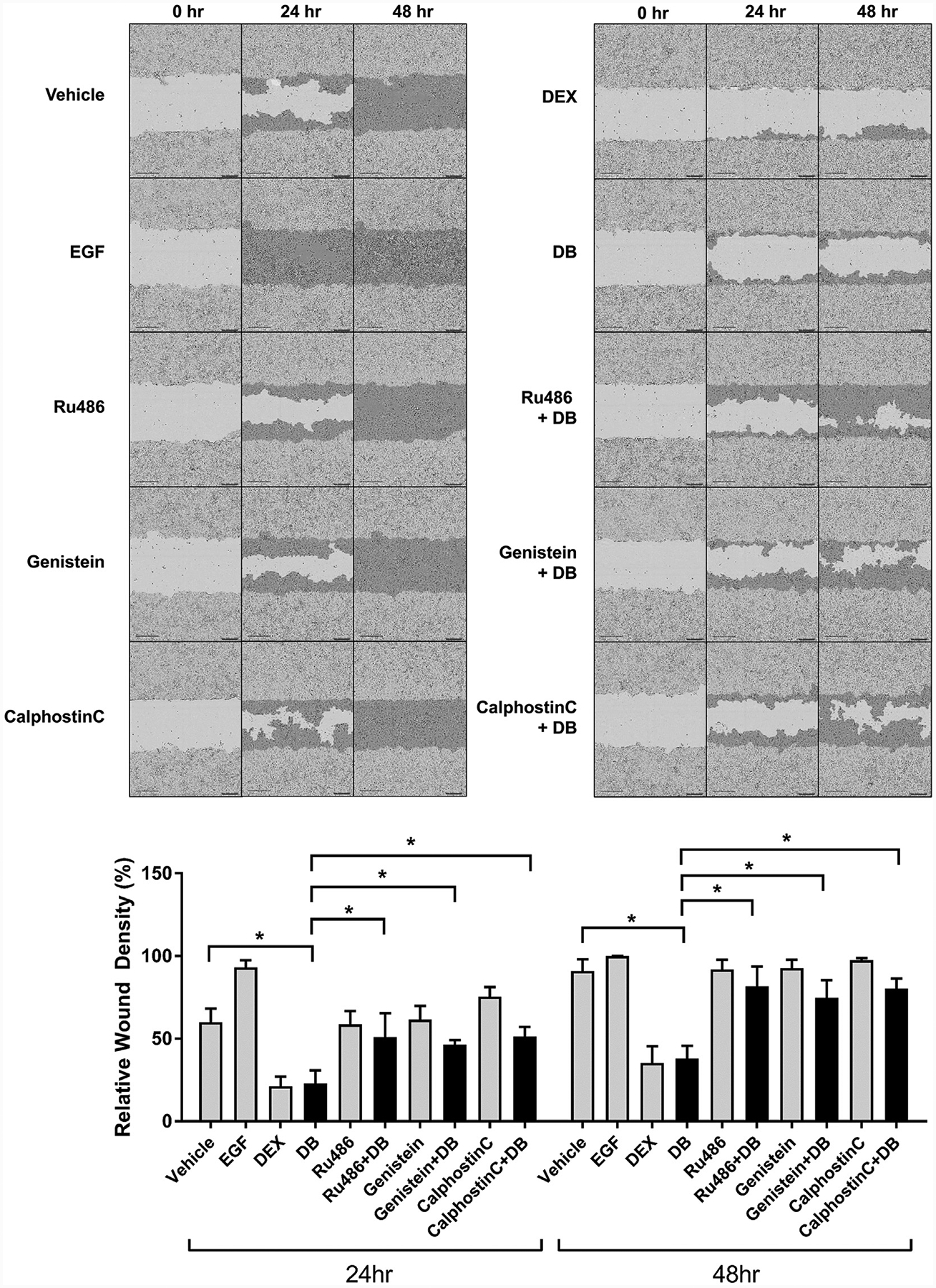
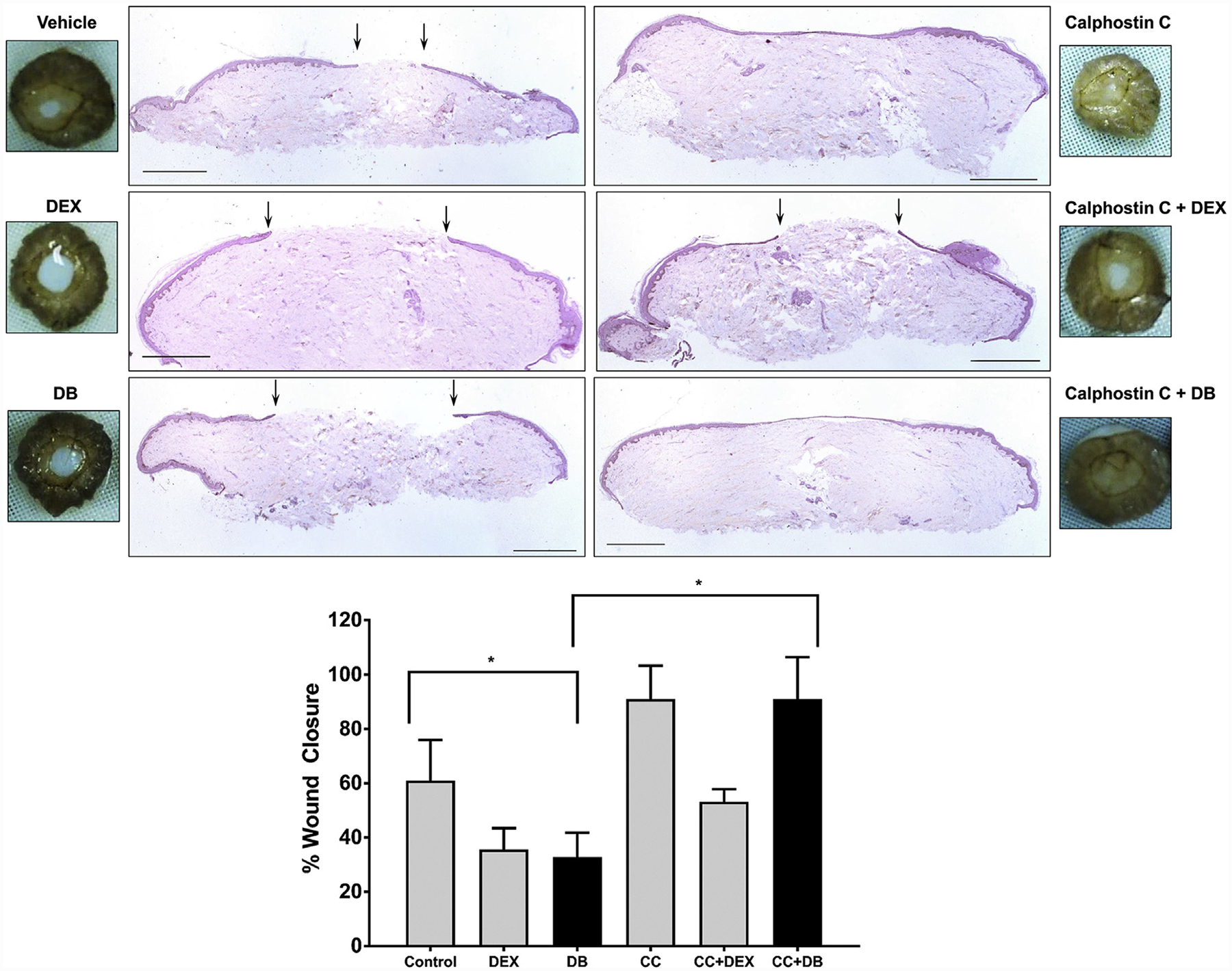
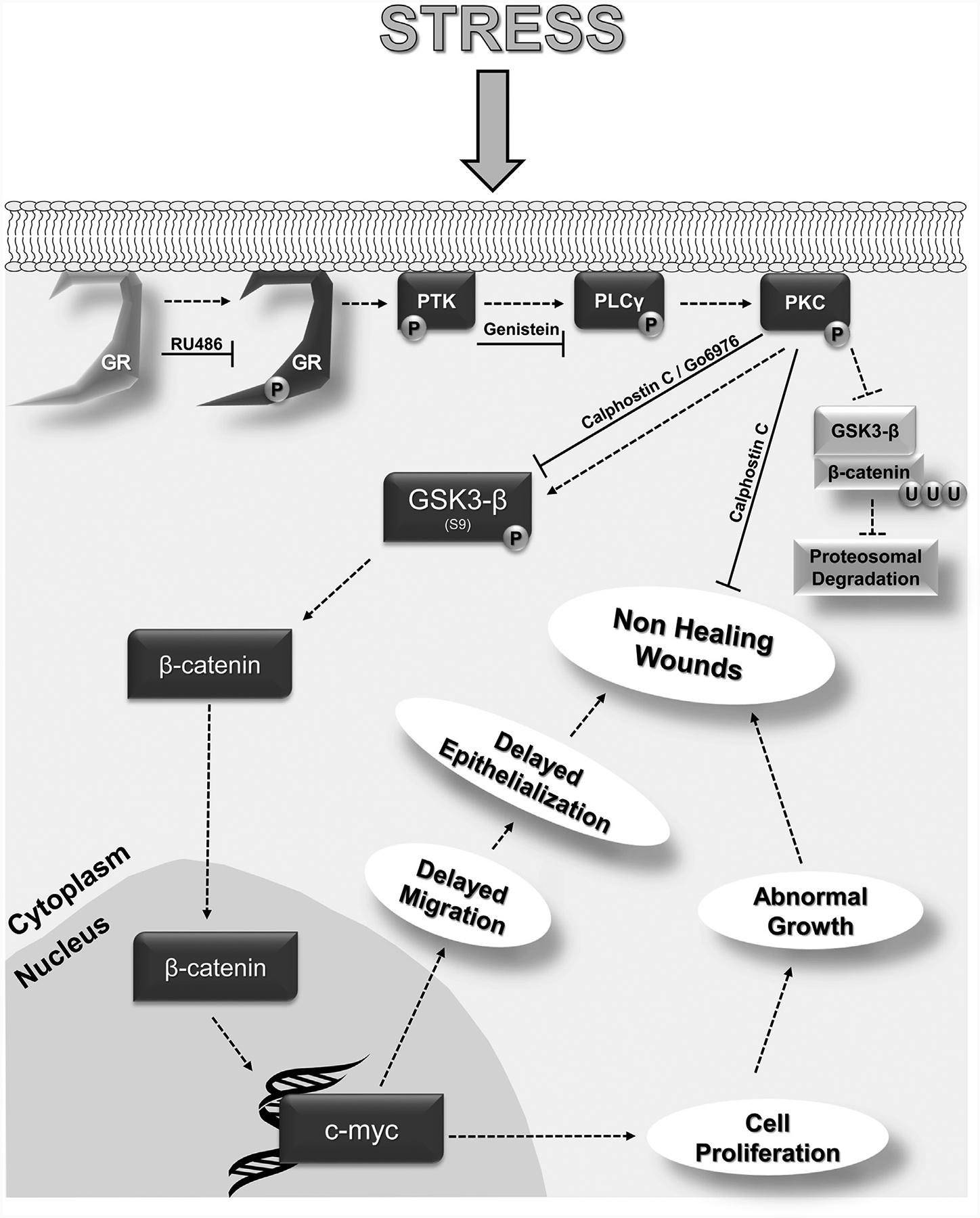
Glucocorticoids (GCs), key mediators of stress signals, are also potent wound healing inhibitors. To understand how stress signals inhibit wound healing, we investigated the role of membranous glucocorticoid receptor (mbGR) by using cell-impermeable BSA-conjugated dexamethasone. We found that mbGR inhibits keratinocyte migration and wound closure by activating a Wnt-like phospholipase (PLC)/ protein kinase C (PKC) signaling cascade. Rapid activation of mbGR/PLC/PKC further leads to activation of known biomarkers of nonhealing found in patients, β-catenin and c-myc. Conversely, a selective inhibitor of PKC, calphostin C, blocks mbGR/PKC pathway, and rescues GC-mediated inhibition of keratinocyte migration in vitro and accelerates wound epithelialization of human wounds ex vivo. This novel signaling mechanism may have a major impact on understanding how stress response via GC signaling regulates homeostasis and its role in development and treatments of skin diseases, including wound healing. To test tissue specificity of this nongenomic signaling mechanism, we tested retinal and bronchial human epithelial cells and fibroblasts. We found that mbGR/PLC/PKC signaling cascade exists in all cell types tested, suggesting a more general role. The discovery of this nongenomic signaling pathway, in which glucocorticoids activate Wnt pathway via mbGR, provides new insights into how stress-mediated signals may activate growth signals in various epithelial and mesenchymal tissues.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
CONFLICT OF INTEREST
Dr. Tomic-Canic is listed as an inventor of a patent PCT/US2010/062361 “Composition and methods for promoting epithelialization and wound closure” issued to the New York University based on the data presented, in part, in the study and stands to potentially gain royalties from future commercialization.
Figures
Comment in
-
Glucocorticoids Inhibit Wound Healing: Novel Mechanism of Action.J Invest Dermatol. 2017 May;137(5):1012-1014. doi: 10.1016/j.jid.2017.01.024. J Invest Dermatol. 2017. PMID: 28411834 Free PMC article.
Similar articles
-
Lucidone Promotes the Cutaneous Wound Healing Process via Activation of the PI3K/AKT, Wnt/β-catenin and NF-κB Signaling Pathways.Biochim Biophys Acta Mol Cell Res. 2017 Jan;1864(1):151-168. doi: 10.1016/j.bbamcr.2016.10.021. Epub 2016 Nov 2. Biochim Biophys Acta Mol Cell Res. 2017. PMID: 27816443
-
Pharmacological and Genetic Inhibition of Caveolin-1 Promotes Epithelialization and Wound Closure.Mol Ther. 2019 Nov 6;27(11):1992-2004. doi: 10.1016/j.ymthe.2019.07.016. Epub 2019 Jul 30. Mol Ther. 2019. PMID: 31409528 Free PMC article.
-
Farnesyl pyrophosphate inhibits epithelialization and wound healing through the glucocorticoid receptor.J Biol Chem. 2010 Jan 15;285(3):1980-8. doi: 10.1074/jbc.M109.016741. Epub 2009 Nov 10. J Biol Chem. 2010. PMID: 19903814 Free PMC article.
-
Drug development targeting the glycogen synthase kinase-3beta (GSK-3beta)-mediated signal transduction pathway: inhibitors of the Wnt/beta-catenin signaling pathway as novel anticancer drugs.J Pharmacol Sci. 2009 Feb;109(2):179-83. doi: 10.1254/jphs.08r28fm. Epub 2009 Jan 29. J Pharmacol Sci. 2009. PMID: 19179804 Review.
-
Molecular insights into diabetic wound healing: Focus on Wnt/β-catenin and MAPK/ERK signaling pathways.Cytokine. 2025 Jul;191:156957. doi: 10.1016/j.cyto.2025.156957. Epub 2025 May 13. Cytokine. 2025. PMID: 40367830 Review.
Cited by
-
The Insulin Receptor: A Potential Target of Amarogentin Isolated from Gentiana rigescens Franch That Induces Neurogenesis in PC12 Cells.Biomedicines. 2021 May 20;9(5):581. doi: 10.3390/biomedicines9050581. Biomedicines. 2021. PMID: 34065446 Free PMC article.
-
The importance of caveolins and caveolae to dermatology: Lessons from the caves and beyond.Exp Dermatol. 2020 Feb;29(2):136-148. doi: 10.1111/exd.14068. Epub 2020 Jan 10. Exp Dermatol. 2020. PMID: 31845391 Free PMC article. Review.
-
Glucocorticoid-mediated induction of caveolin-1 disrupts cytoskeletal organization, inhibits cell migration and re-epithelialization of non-healing wounds.Commun Biol. 2021 Jun 18;4(1):757. doi: 10.1038/s42003-021-02298-5. Commun Biol. 2021. PMID: 34145387 Free PMC article.
-
Glucocorticoids Inhibit Wound Healing: Novel Mechanism of Action.J Invest Dermatol. 2017 May;137(5):1012-1014. doi: 10.1016/j.jid.2017.01.024. J Invest Dermatol. 2017. PMID: 28411834 Free PMC article.
-
Notch1 signaling determines the plasticity and function of fibroblasts in diabetic wounds.Life Sci Alliance. 2020 Oct 27;3(12):e202000769. doi: 10.26508/lsa.202000769. Print 2020 Dec. Life Sci Alliance. 2020. PMID: 33109684 Free PMC article.
References
-
- Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol 2002;71:9–15. - PubMed
-
- Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J 2004;18: 70–80. - PubMed
-
- Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, et al. Protein kinase Cdelta mediates insulin-induced glucose transport in primary cultures of rat skeletal muscle. Mol Endocrinol 1999;13:2002–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
