

DESCARTES' RULE OF SIGNS AND THE IDENTIFIABILITY OF POPULATION DEMOGRAPHIC MODELS FROM GENOMIC VARIATION DATA
- PMID: 28018011
- PMCID: PMC5175586
- DOI: 10.1214/14-AOS1264
DESCARTES' RULE OF SIGNS AND THE IDENTIFIABILITY OF POPULATION DEMOGRAPHIC MODELS FROM GENOMIC VARIATION DATA
Abstract
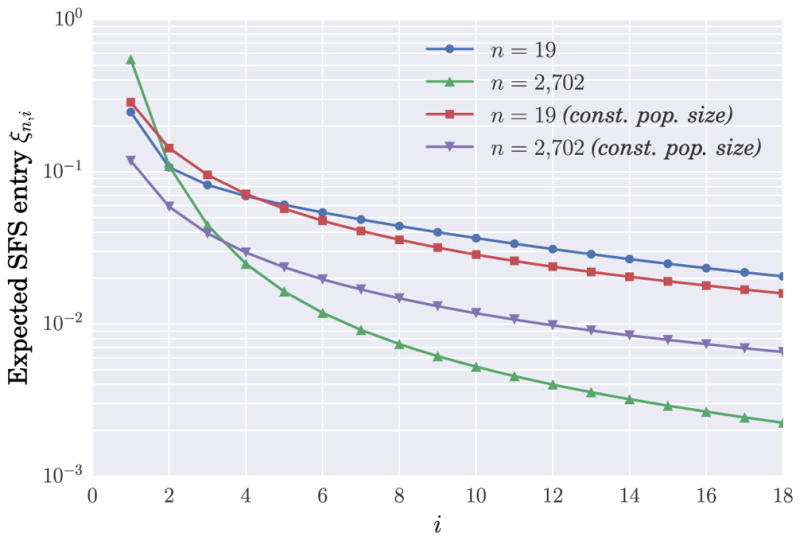
The sample frequency spectrum (SFS) is a widely-used summary statistic of genomic variation in a sample of homologous DNA sequences. It provides a highly efficient dimensional reduction of large-scale population genomic data and its mathematical dependence on the underlying population demography is well understood, thus enabling the development of efficient inference algorithms. However, it has been recently shown that very different population demographies can actually generate the same SFS for arbitrarily large sample sizes. Although in principle this nonidentifiability issue poses a thorny challenge to statistical inference, the population size functions involved in the counterexamples are arguably not so biologically realistic. Here, we revisit this problem and examine the identifiability of demographic models under the restriction that the population sizes are piecewise-defined where each piece belongs to some family of biologically-motivated functions. Under this assumption, we prove that the expected SFS of a sample uniquely determines the underlying demographic model, provided that the sample is sufficiently large. We obtain a general bound on the sample size sufficient for identifiability; the bound depends on the number of pieces in the demographic model and also on the type of population size function in each piece. In the cases of piecewise-constant, piecewise-exponential and piecewise-generalized-exponential models, which are often assumed in population genomic inferences, we provide explicit formulas for the bounds as simple functions of the number of pieces. Lastly, we obtain analogous results for the "folded" SFS, which is often used when there is ambiguity as to which allelic type is ancestral. Our results are proved using a generalization of Descartes' rule of signs for polynomials to the Laplace transform of piecewise continuous functions.
Keywords: Population genetics; Primary 62B10; coalescent theory; frequency spectrum; identifiability; population size; secondary 92D15.
Figures
References
-
- Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. Demonstrating stratification in a European American population. Nat Genet. 2005;37:868–872. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources